Download
1 / 32
320 likes | 336 Views
Explore popular methods for finding exons in protein coding genes, from ab initio predictions to cDNA sequencing. Learn the challenges and advantages of different approaches in gene prediction.

E N D
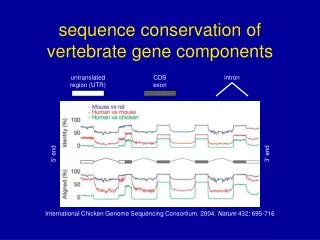
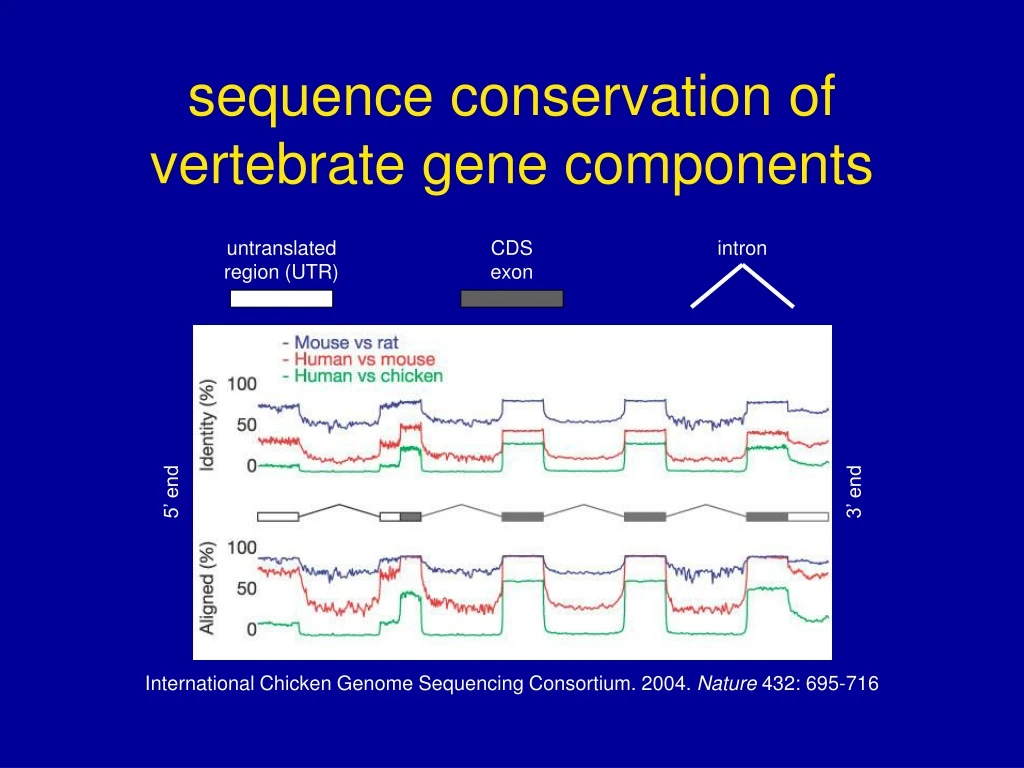
untranslated region (UTR) CDS exon intron 5’ end 3’ end International Chicken Genome Sequencing Consortium. 2004. Nature 432: 695-716 sequence conservation of vertebrate gene components
popular methods for finding exons in protein coding genes • ab initio computer predictions • PRO: can identify genes expressed at low levels or under rare conditions • CON: tradeoffs between false positives and negatives • reverse transcribe mRNA into cDNA and sequence • PRO: the “gold standard” even if getting full length cDNAs is problematic • CON: genes expressed at low levels or under rare conditions are missed • hybridize cDNA to tiling array • PRO: no need to wade through highly expressed genes • CON: genes expressed at low levels or under rare conditions are missed • CON: determining start/end of transcript is problematic
16 of the largest human genesbased on cDNA alignments to BAC-end consistent genomic contigs
16 of the largest human intronsbased on cDNA alignments to BAC-end consistent genomic contigs
3.6 Mb intron full of microsatellites Reugels AM, et al. Genetics154: 759-769 (2000) 3.6 Mbp intron in the dynein gene DhDhc7(Y) on the heterochromatic Drosophila hydei Y chromosome
based on estimates published in Wong GK, et al. 2001. Most of the human genome is transcribed. Genome Res 11: 1975-1977 how much transcribed DNA is attributable to genes over 100 Kb
most exons are 150 bp except for the 3’ terminal exon with the UTR large genes are attributable to more introns and to bigger introns
information used by ab initio algorithms for exon prediction • signal terms = short sequence motifs like splice sites, branch points, polypyrimidine tracts, start codons, and stop codons • is almost enough to define the genes when the introns are small like in yeast • BUT is not adequate when the introns are large like in human • content terms = patterns of codon usage that are unique to a species, and optionally, cross species sequence conservation • algorithm must be trained by presenting them with known coding sequences • caution 1: untranslated regions (UTR)s cannot be detected • caution 2: non-protein-coding RNA genes cannot be detected • caution 3: alternatively spliced isoforms are not considered • ab initio method is used when there is no full length cDNA (or protein)
cDNA or protein in another species false positives but false gene deserts reject unmatched exons novel genes genome sequence ab initio prediction reject unmatched genes putative genes EST in the chosen species not in final gene counts based on Curwen V, … Clamp M (2004) Genome Res 14: 942 but modified according to reviews by Wang J, … Wong GK (2003) Nat Rev Genet 4: 741 Ensembl process in absence of full length cDNA (or protein)
false gene desert over-prediction false positive gene fragment Refseq = full length cDNA; Genscan/FgeneSH = ab initio algorithms; Ensembl = final annotation example of what can go wrong in transition from ab initio to Ensembl
Genscan prediction fails at both extremes in size; lower sizes correspond to single-exon genes; upper sizes are due to large introns Ensembl does everything to minimize the FP rate but in doing so it increases the FN rate to almost 50% size dependencies of FP and FN
over-predictions arise when the ab initio algorithms fail to detect the start and stop codons at the ends of a gene; most performance assessments confuse this issue with FP but it is a distinct phenomenon because unlike FP the probability of an over-prediction is independent of size in contrast to FP and FN, over-predictions are size independent
a complete miss (CM) is a gene where fewer than 100 bp of the total protein coding sequence is correctly predicted a false desert (FD) is the fraction of a gene’s sequence that is not covered by any gene predictions; notice that definition of FD must exclude CM genes false gene deserts from Ensembl
gene fragments Refseq is a curated set of full length cDNAs; the right panel shows what is left after removing cDNA derived genes from Ensembl (human-32.35e 7/21/2005) gene size distribution with and without full length cDNA support
ENCODE Project Consortium. 2007. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447: 799-816 [excerpt of abbreviations from box 1] CDS Coding sequence: a region of a cDNA or genome that encodes proteins CS Constrained sequence: a genomic region associated with evidence of negative selection (that is, rejection of mutations relative to neutral regions) GENCODE Integrated annotation of existing cDNA and protein resources to define transcripts with both manual review and experimental testing procedures PET A short sequence that contains both the 5' and 3' ends of a transcript RACE Rapid amplification of cDNA ends: a technique for amplifying cDNA sequences between a known internal position in a transcript and its 5' end RxFrag Fragment of a RACE reaction: a genomic region found to be present in a RACE product by an unbiased tiling-array assay TxFrag Fragment of a transcript: a genomic region found to be present in a transcript by an unbiased tiling-array assay Un.TxFrag A TxFrag that is not associated with any other functional annotation UTR Untranslated region: part of a cDNA either at the 5' or 3' end that does not encode a protein sequence
Shoemaker DD, et al. 2001.Nature 409: 922-927 [nonrepetitive half of human genome requires 150 million probes if tiled at 10 bp steps] weakness of the method is it cannot determine the ends of the gene experimental annotation of a genome using tiling microarrays
most TxFrags (63.5%) do not concur with the GENCODE exons and are observed in intronic (40.9%) and intergenic (22.6%) regions; annotated TxFrags are more likely to be seen in multiple cell lines; more disturbingly these unannotated TxFrags contain little evidence of encoding proteins annotated and unannotated TxFrags vs number of cell lines
RACE (rapid amplification of cDNA ends) is a way to get the ends of a gene by priming off the incomplete cDNA; using 399 protein-coding loci and mRNA for 12 tissues they found that 90% of these loci contain at least one novel RxFrag that extends well beyond the annotated TSS extension of annotated genes based on the RACE experimentsmean gene size was 27 kb in the 2001 human genome papers
330-kb interval of human chromosome 21 with 4 annotated genes: DONSON, CRYZL1, ITSN1 and ATP5O; 5’ RACE products generated from small intestine RNA and detected by tiling-array analyses (RxFrags) are shown along the top; magnified along the bottom is a cloned and sequenced RT–PCR product with 2 exons from the DONSON gene and 3 exons from the ATP5O gene connected by a single large 300 kb intron; PET tags show the termini of a transcript that is consistent with this RT–PCR product in fact approximately 50% of RACE-positive loci appear to have incorporated at least 1 exon from an upstream gene multiple lines of evidence for the fusion of two adjacent genes
GENCODE annotations, RACE-array experiments, and PET tags were used to assess the presence of a nucleotide in a primary transcript; the proportion of genomic bases detected can be classified into the following scenarios: all three technologies, two of the three technologies, one technology but with multiple observations, and one technology with only one observation; also indicated are genomic bases without any detectable coverage of primary transcripts most of the human genome is converted into primary transcripts
ENCODE confirmed previous studies in human and mouse showing extensive transcription beyond the official annotations93% of bases are represented in a primary transcript identified by at least 2 independent observations, some by same technologymany of the resulting transcripts are neither traditional protein-coding genes nor explainable by structural non-coding RNAsthe rest of the paper shows extensive amounts of regulatory factors around the novel transcription start sites, as is to be expectedcompared to other annotated features unannotated transcripts show weaker (i.e. almost neutral) evolutionary conservationbiological relevance of unannotated transcripts remains unanswered
Thomas JW, et al. 2003.Comparative analyses of multi-species sequences from targeted genomic regions. Nature 424: 788-793 Q: what is the optimal species or combination of species to use? conservation patterns comparing human to other vertebrate genomes
evolutionarily constrained regions are computed for 28 vertebrate species and defined to have a false discovery rate of 5%; the median length of the constrained sequences is 19 bp, and the minimum length is 8 bp or about the size of a typical transcription factor binding site evolutionarily constrained regions are not always ENCODE annotated
increase in significance from bases to regions definition is indication that tiny islands of constrained sequences exist in the experimentally defined functional elements whereby the surrounding bases seem not to be constrained ENCODE annotated regions are not always evolutionarily constrained
identifying functional elements from genome sequence is one challenge, but what biological roles (if any) do the elements serve?sequence similarity to previously characterized genes and proteins is commonly used to infer biological roles, but no one has ever quantified how reliable these inferences might beascertainment of biological roles is extremely difficult as most knockouts have no phenotype even for indisputably reliable genes
S1 ortholog B1 paralog paralog S2 B2 ortholog species S species B http://www.treefam.org/ is a curated database of animal gene family trees with reliable assignments of ortholog and paralog does orthology necessarily imply functional equivalence?
Benner SA, Gaucher EA. 2001. Trends Genet 17: 414-418 Homologous enzymes catalyze four different reactions that are involved in (a) central metabolism, i.e. the citric acid cycle (b) amino acid degradation (c) nucleic acid biosynthesis and (d) amino acid biosynthesis. There is NO question that the four enzymes are homologous, but their biological roles are arguably quite different. evolution, language, and analogy in functional genomics
2.83 million variant sites chicken SNP map chicken genome 1065 human genes taken from OMIM 1 cSNP intolerant in SIFT 520 cSNPs in 245 genes 995 chicken orthologs 6 cSNPs in disease site 5 cSNPs tolerant in SIFT if orthologs are functionally equivalent no SNPs would survive the process but a few do in paper of Wong GK, … Yang H. 2004. Nature 432: 717-722 chicken SNPs corresponding to mutations in human disease genes
ornithine transcarbamylase (OTC) 188 Human HYSSLKGLTLSWIGDGN Pig --GA------------- Mouse --G-------------- Rat --G-------------- Chicken RJF --GG-N---IA------ Chicken B/L --GG-NR--IA------ mutation associated with hyperammonemia in humans turns out to be a common polymorphism in healthy chickens, with the deleterious variant observed in 65% of layers and 75% of broilers G188R substitution associated is with hyperammonemia in humans
mammals UREA waste; birds-and-reptiles URIC acid waste every human urea cycle gene (including OTC) is found in chicken Q: could we have predicted OTC’s lack of functional equivalence? nitrogenous waste processing in mammals versus birds-and-reptiles
2NeΔt > 1 / Δw Ne is effective population size Δt is number of generations while Δw is differential fitness if we think of one generation for one member of a population as a single evolutionary experiment, then we can never hope to duplicate the number of experiments that nature conducted in order to decide what will survive “genetic uncertainty principle” explains why a gene’s biological role is so difficult to ascertainhypothesis by Tautz D. 2000. Trends Genet 16: 475-477