Download

1 / 21
300 likes | 556 Views
Short read mapping (Alignment). Alignment topics in GEN875. Whole genome alignment Short read “mapping” BLAST Pair-wise using dynamic programming Progressive multiple alignment. Alignment. Take a set of sequences. Find where they match.

E N D
Alignment topics in GEN875 • Whole genome alignment • Short read “mapping” • BLAST • Pair-wise using dynamic programming • Progressive multiple alignment
Alignment • Take a set of sequences. Find where they match. • Arrange sequences in a matrix where columns contain homologous (corresponding?) characters from each sequence
Types of Alignments • Global – include the entire length of all sequences in the alignment • Local – identify and align subsets of longer sequences • Glocal - hybrid
Short Read Mapping • Find a match between sequence reads and a reference genome • Find the best match between sequence reads and a reference genome • Find all the plausible matches between sequence reads and a reference genome
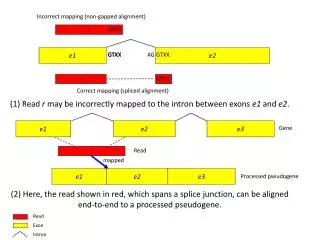
Reads may not match the reference exactly • Sequence errors in the read – may be distinguishable using quality scores • Sequence errors in the reference genome • Legitimate polymorphism
Phred Scores Phred Score P( incorrect base ) Base call accuracy 10 1 in 10 90% 20 1 in 100 99% 30 1 in 1000 99.9% 40 1 in 10000 99.99% 50 1 in 100000 99.999%
@SEQ_ID GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT + !''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65 Sanger format encodes a Phred quality score from 0 to 93 using ASCII 33 to 126 Illumina uses several variants
When (how much) does (sequence and ) alignment accuracy matter? • RNA seq for expression • RNA seq for annotation – endpoints, splicing • chIP seq • Resequencing related genomes for SNP detection • Resequencing related genomes for indel detection • Resequencing to clean up existing sequences • Sequencing to determine copy number
Short Read Mapping Tools • Bowtie • ELAND (Illumina) • Maq • SOAP • RMAP • ZOOM • SHRiMP • BFAST • MOSAIK • BWA • SOAP2 • Speed • Accuracy • Exact match vs mismatches • Gapped vs ungapped • Greedy or exhaustive
Short Read Mapping Tools • Bowtie • ELAND (Illumina) • Maq • SOAP • RMAP • ZOOM • SHRiMP • BFAST • MOSAIK • BWA • SOAP2 • Hash table of oligos in reference sequence • Hash table of input reads • Hash table – method unknown • Burrows Wheeler Transform-based Index
Spaced Seeds Example Seed: 1100 Query: GATC Matches: GATC GAAC GACC GATT GATA GATG • Length and weight of seeds • Number of Hash tables required to find mismatches
Some mapping software uses alignment refinement • Once candidates are identified using the hash table search, conduct a more rigorous alignment of the read and reference genome • Smith-Waterman local alignment (with or without gaps)
Bowtie • Burrows Wheeler Index based on FM index extended to accommodate mismatches • Reduces memory footprint • Increases speed • Amenable to multiple processors • 14 .3x Illumina coverage of human genome mapped in 14 hrs on a 4 core desktop PC
Query Sequence: GGTA No exact match, so try alternative sequences with a mismatch: GGCA GGAA GGTG
Bowtie caveat “If one or more exact matches exist for a read, then Bowtie is guaranteed to report one, but if the best match is an inexact one then Bowtie is not guaranteed in all cases to find the highest quality alignment.” …unless you use the slower “best” option