Download

1 / 29
290 likes | 423 Views
Short read mapping (Alignment). ABySS:. Kmer n n:100 n:N50 min median mean N50 max sum 20 11465 4945 862 100 686 1084 1891 17871 5364700 25 6206 2521 278 100 1026 2138 4578 36808 5390722 30 4887 2134 181 100 998 2577 7525 62225 5500829 35 4261 1945 159 100 1002 2831 9493 66466 5507159

E N D
ABySS: Kmer n n:100 n:N50 min median mean N50 max sum 20 11465 4945 862 100 686 1084 1891 17871 5364700 25 6206 2521 278 100 1026 2138 4578 36808 5390722 30 4887 2134 181 100 998 2577 7525 62225 5500829 35 4261 1945 159 100 1002 2831 9493 66466 5507159 40 3693 1827 147 101 1049 3089 10663 69602 5645100 45 3727 1797 145 100 1006 3062 10791 71989 5502746 50 27051791145 100 1053 3069 10378 82440 5496994 55 3288 2636 148 100 398 2097 9959 71517 5528981 60 3053 2531 150 100 459 2242 10487 77123 5674674 64 3001 2571 164 100 499 2157 9508 47608 5546956 Newbler: n n:100 n:N50 min median mean N50 max sum 298 298 37 100 6747 18245 45215 185235 5437024 Enterobacter cloacae subsp. cloacae ATCC 13047 chromosome, Length: 5,314,581 nt plasmid pECL_B, Length: 84,653 nt plasmid pECL_A, Length: 199,562 nt Total Length: 5598796
Alignment topics in GEN875 • Assembly • Whole genome alignment • Short read “mapping” • BLAST • Pair-wise using dynamic programming • Progressive multiple alignment
Alignment • Take a set of sequences. Find where they match. • Arrange sequences in a matrix where columns contain homologous (corresponding?) characters from each sequence
Types of Alignments • Global – include the entire length of all sequences in the alignment • Local – identify and align subsets of longer sequences • Glocal - hybrid
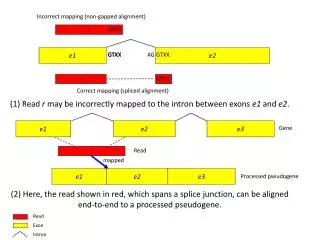
Short Read Mapping • Find a match between sequence reads and a reference genome • Find the best match between sequence reads and a reference genome • Find all the plausible matches between sequence reads and a reference genome
Reads may not match the reference exactly • Sequence errors in the read – may be distinguishable using quality scores • Sequence errors in the reference genome • Legitimate polymorphism
When (how much) does (sequence and ) alignment accuracy matter? • RNA seq for expression • RNA seq for annotation – endpoints, splicing • chIP seq • Resequencing related genomes for SNP detection • Resequencing related genomes for indel detection • Resequencing to clean up existing sequences • Sequencing to determine copy number
Short Read Mapping Tools • Bowtie • ELAND (Illumina) • Maq • SOAP • RMAP • ZOOM • SHRiMP • BFAST • MOSAIK • BWA • SOAP2 • Speed • Accuracy • Exact match vs mismatches • Gapped vs ungapped • Greedy or exhaustive
Short Read Mapping Tools • Bowtie • ELAND (Illumina) • Maq • SOAP • RMAP • ZOOM • SHRiMP • BFAST • MOSAIK • BWA • SOAP2 • Hash table of oligos in reference sequence • Hash table of input reads • Hash table – method unknown • Burrows Wheeler Transform-based Index
Spaced Seeds Example Seed: 1100 Query: GATC Matches: GATC GAAC GACC GATT GATA GATG • Length and weight of seeds • Number of Hash tables required to find mismatches
Some mapping software uses alignment refinement • Once candidates are identified using the hash table search, conduct a more rigorous alignment of the read and reference genome • Smith-Waterman local alignment (with or without gaps)
Bowtie • Burrows Wheeler Index based on FM (full-text minute) index extended to accommodate mismatches • Reduces memory footprint • Increases speed • Amenable to multiple processors • 14 .3x Illumina coverage of human genome mapped in 14 hrs on a 4 core desktop PC
gc$aaac $ a c g 0 1 4 6 For each character, make a table of the number of lexicographically smaller characters in the text For each position, make a matrix of the number of occurrences of each character in the prefix LF mapping is simple addition
Query Sequence: GGTA No exact match, so try alternative sequences with a mismatch: GGCA GGAA GGTG
Bowtie caveat “If one or more exact matches exist for a read, then Bowtie is guaranteed to report one, but if the best match is an inexact one then Bowtie is not guaranteed in all cases to find the highest quality alignment.” …unless you use the slower “best” option
Alignment Methods – Dynamic Programming • Needleman-Wunsch (global) and Smith-Waterman (local) use dynamic programming • Guaranteed to find an optimal alignment given a particular scoring function • Computationally intensive
Dynamic Programming • One possible simple scoring scheme: • Si,j = 1 if the residue at position i of sequence #1 is the same as the residue at position j of sequence #2 (match score); otherwise • Si,j = 0 (mismatch score) • w = 0 (gap penalty)
Dynamic Programming Three steps: 1) Initialize 2) Fill Matrix Mi,j = MAXIMUM [Mi-1, j-1 + Si,j (match/mismatch in the diagonal), Mi,j-1 + w (gap in sequence #1), Mi-1,j + w (gap in sequence #2)]
Dynamic Programming 3) Traceback G A A T T C A G T T A G G A - T C - G - - A Score = 1+0+1+0+1+1+0+1+0+0+1 = 6