Download

1 / 58
580 likes | 599 Views
Amyloidosis for the young ladies. Amyloidosis.

E N D
Amyloidosis Definition : In medicine, amyloidosis refers to a variety of conditions in which amyloidproteins are abnormally deposited in organs causing disease. A protein is amyloid if, due to an alteration in its secondary structure, it takes on a particular insoluble form, called the beta-pleated sheet.
AMYLOIDOSIS Disease characterized by extracellular deposition of pathologic insoluble fibrillar proteins in organs and tissues. Term amyloid first coined by Virchow in mid 19th century (meaning starch or cellulose). Amyloid found to stain with congo red, appearing red microscopically in normal light but apple green when viewed in polarized light. Fibrillar nature and beta pleated sheet configuration described by electron microscopy in 1959.
Protein Misfolding Diseases • A specific protein may be unable to carry out its normal function because it is improperly folded or because it is not sufficiently stable due to misfolding. • A protein may be unable to carry out its normal function because it is not trafficked to the proper location due to misfolding. • A protein may fail to fold or to remain folded correctly and consequently aggregate (often with other components) leading to amyloid diseases. (“Amyloidosis” refers strictly to diseases in which extracellular deposits are formed, but the terms “amyloid diseases” or “protein aggregation diseases” are now being used to refer to diseases in which protein deposits are intra- or extracellular.) • Some of the clinical symptoms of the non-neurological amyloidoses seem to be due to the accumulation of large deposits of aggregated proteins in vital organs • In neurodegenerative diseases, cellular function appears to be impaired by the interaction of aggregated proteins with cellular components. This impairment is associate with evidence of elevated oxidative stress, but the mechanism is unknown.
Systemicamyloidoses are those which affect more than one body organ or system. Localisedamyloidoses affect only one body organ or tissue type. Primaryamyloidoses arise from a disease with disordered immune cell function such as multiple myeloma and other immunocytedyscrasias. Secondary (reactive) amyloidoses are those occurring as a complication of some other chronic inflammatory or tissue destructive disease.
Imaging techniques – Technetium Tc 99m pyrophosphate binds avidly to many types of amyloid. Quantitative assessment not possible and strongly positive results usually only occur in pt’s with severe disease. Technetium labeled aprotinin may be more sensitive. • Quantitative scintigraphy can be done with iodine-123- labeled serum amyloid P component (sensitive for AL, ATTR and AA amyloid).
Multiple Myeloma: Incidence and Etiology 13,000 cases/year in USA Median age - 65 yrs. Incidence in African-Americans is two-fold other ethnic groups Familiar clusters are rare Environmental/occupational exposures have been implicated
AL AMYLOIDOSIS Part of the spectrum of plasma cell dyscrasias. Cellular source of AL amyloid is always a single clone of the B-lymphocytic lineage, usually exhibiting the morphologic appearance of plasma cells. Underlying clonal proliferative disorder may be frankly neoplastic (ie:multiple myeloma) or conversely a low grade proliferation of monoclonal plasma cells.
Heart failure Autonomic nervous system involvement Macroglossia hoarsenes Carpal tunnel sy Peripheral nervous system involvement Thrombocytosis AL AMYLOID Splenomegaly Anemia Hypofunction of adrenal glands hypothyreosis Hepatomegaly Nephrotic syndrome
Among MM patients, amyloidosis reported with variable frequency, but rarely exceeds 20%. Majority of patients without myeloma associated AL, occurs in the setting of an apparently “benign” monoclonal gammopathy. Characterized by low concentrations of monoclonal Ig’s in serum/urine and an often occult low grade monoclonal plasma cell proliferation in BM.
AL Amyloidosis IF anti l Kidney (74%) Heart (58%) Bone marrow plasma cell clone synthesizing amyloidogenic light chain Liver (28%) No of organs involved: 1 (31%) >1 (69%) GI (8%)
Presenting Clinical Features in AA Amyloidosis Feature % Proteinuria/renal insufficiency 91 Diarrhea/malabsorption 22 Goiter 9 Neuropathy/carpal tunnel syndrome 3 Hepatomegaly 5 Lymphadenopathy 2 Cardiac 1 - 2
Macroglossia – occurs in 10-20 % Amyloid can be found within any part of the GI tact and may infiltrate parenchyma, organs and nerves. Peripheral neuropathy may be presenting manifestation or develop subsequently during the course of the illness (history of carpal tunnel frequently elicited). Neuropathy usually distal, symmetric and progressive. Cranial nerve and autonomic nerve involvement also well described. Motor neuropathy rare.
HEPATIC/SPLENIC Involvement of liver common. Hepatomegaly may be striking at presentation and usually disproportionate to extent of liver enzyme abnormalities (except alkaline phosphatase which is frequently elevated). Presence of jaundice is an adverse prognostic factor and MST from onset of jaundice is only 3 months. Patients may present with severe intrahepatic cholestasis. Massive splenic deposition may result in functional hyposplenism.
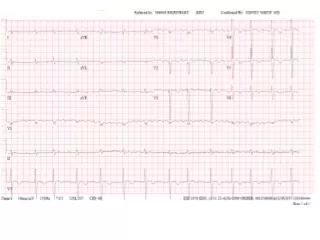
CARDIAC May present with rapid and progressive onset of CHF. Characteristically, features are predominantly of right sided CHF. ECG – low voltage and may have a pattern of MI in absence of CAD. ECHO – concentrically thickened ventricles with normal-small cavity and diastolic dysfunction on doppler. Clinical clue is marked worsening of failure when CCB used.
Echocardiogram revealing thickened walls with small chambers
RENAL Nephrotic syndrome present in 30-50% at diagnosis. Nephrotic syndrome and renal failure develop only rarely during course of the illness if not present at time of diagnosis. λ BJP have been associated with inferior survival as compared with κBJP or no monoclonal protein, irrespective of serum creatinine.
Lambda Kappa