Download
1 / 26
370 likes | 1.16k Views
Cardiac Amyloidosis. Ann Isaksen Morning Report November 10, 2009. Causes of Non-Ischemic Cardiomyopathy. Infiltrative (Sarcoidosis, Amyloidosis, Hemocromatosis) Viral (HIV, lyme, coxsackie, etc) Endocrine (Thyroid, pheo, cushing’s) SLE Drug/toxin induced (EtOH, cocaine, arsenic,chemo)

E N D
Cardiac Amyloidosis Ann Isaksen Morning Report November 10, 2009
Causes of Non-Ischemic Cardiomyopathy • Infiltrative (Sarcoidosis, Amyloidosis, Hemocromatosis) • Viral (HIV, lyme, coxsackie, etc) • Endocrine (Thyroid, pheo, cushing’s) • SLE • Drug/toxin induced (EtOH, cocaine, arsenic,chemo) • Nutritional deficiencies (thiamine, selenium) • Malignancy • Pregnancy • ?Celiac disease
Amyloidosis • Rudolph Virchow in 1854 adopted the term "amyloid“ to refer to tissue deposits of material that stained in a similar manner to cellulose when exposed to iodine • Amyloidosis is a generic term that refers to the extracellular tissue deposition of fibrils composed of low molecular weight subunits (most of which are in the molecular weight range of 5 to 25 kD) of a variety of proteins. • At least 25 different human and eight different animal protein precursors of amyloid fibrils are now known • “Apple-green birefringence”
Many kinds of Amyloidosis • Primary (AL amyloidosis) • = plasma cell dyscrasia leading to overproduction of Immunoglobulin light chains • Clinical evidence of cardiac involvement occurs in up to 50 percent of patients • Secondary (AA amyloidosis) • Deposition of fragments of serum amyloid A protein, an acute phase reactant • Associated with chronic inflammatory disorders (eg RA). • Almost never produces clinically apparent heart disease (< 5%) • Senile systemic and Heritable amyloidosis • = Transthyretin deposits • + Cardiac involvement, but much slower time course than AL
Many kinds of amyloidosis • Primary (AL amyloidosis) • = plasma cell dyscrasia leading to overproduction of Immunoglobulin light chains • Clinical evidence of cardiac involvement occurs in up to 50 percent of patients • Secondary (AA amyloidosis) • Deposition of fragments of serum amyloid A protein, an acute phase reactant • Associated with chronic inflammatory disorders (eg RA). • Almost never produces clinically apparent heart disease (< 5%) • Senile systemic and Heritable amyloidosis • = Transthyretin deposits • +Cardiac involvement, but much slower time course than AL
Clinical Manifestations of AL amyloidosis • Nephrotic syndrome with or without renal insufficiency • Peripheral neuropathy, typically axonal, which can be associated with autonomic neuropathy. Carpal tunnel syndrome is commonly seen • Hepatomegaly, with elevated liver enzyme levels • Macroglossia • Purpura, characteristically elicited in a periorbital distribution (raccoon eyes) by a valsalva maneuver or minor trauma, is present in only a minority of patients, but is highly characteristic of AL amyloidosis
Cardiac exam findings • Elevation of the jugular venous pressure, sometimes with a low-volume pulse. • Right sided heart failure hepatomegaly and LE edema • A right-sided third heart sound is occasionally heard • Fourth heart sound, which coincides with atrial systole, argues against the diagnosis since atrial infiltration causes atrial dysfunction • Amyloidosis rarely causes significant valve disease, but a murmur of tricuspid or mitral regurgitation is occasionally heard.
Diagnostic Evaluation • ECG • TTE • Cardiac MRI • Tissue biopsy • SPEP/UPEP
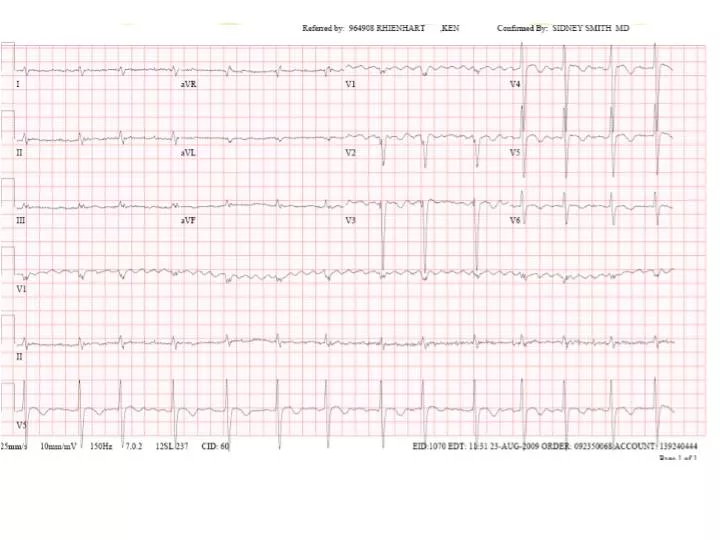
ECG Findings • The most common abnormality = low voltage in the limb leads • Occurs in approximately 50 percent of patients • Other changes that can occur include • 1st degree AV block (21%) • atrial fibrillation or flutter (20%) • Non-specific intraventricular conduction delay (16%) • VTach (5%) • 2nd or 3rd degree AV block (3%)
Echocardiography • Left ventricular wall thickening with evidence of diastolic dysfunction is the earliest echocardiographic abnormality, • In more advanced disease, wall thickening progresses resulting in a restrictive cardiomyopathy with a nondilated or small LV cavity, biatrial enlargement • Amyloid infiltration of the heart results in increased echogenicity. • "granular, sparkling" appearance of the myocardium, unusually high quality myocardial visualization • Only a minority of patients has this pattern 26% = low sensitivity
Two-dimensional (2D) echocardiographic image (parasternal long-axis view) from a patient with AL cardiac amyloidosis showing normal biventricular dimensions, granular "sparkling" ventricular wall appearance, concentric left ventricular wall thickening, and thickened mitral valve leaflets suggesting infiltration.
Voltage-to-mass ratio • Left ventricular thickening due to amyloid infiltration may be misdiagnosed as left ventricular hypertrophy. • However, unlike true left ventricular hypertrophy, left ventricular thickening in cardiac amyloidosis is associated with a decrease in electrocardiographic voltage. • This combination of increased ventricular mass with reduced electrocardiographic voltage is unique to infiltrative cardiomyopathy. • In another report, the combination of low voltage on ECG and an interventricular septal thickness >1.98 cm detected amyloidosis with a sensitivity and specificity of 72 and 91 percent, respectively
Cardiac MRI • Amyloidosis global and subendocardial late gadolinium enhancement (LGE) of the myocardium. • Replacing Echo as imaging modality of choice in pt’s whom you have high clinical suspicion for amyloid cardiomyopathy
Monoclonal Paraprotein • SPEP • Monoclonal Lambda or Kappa Light chain spike • Free serum light chains • The presence of a serum or urine monoclonal paraprotein is suggestive of AL amyloidosis, but it alone does not firmly establish the diagnosis. • Pt may have senile cardiac amyloid and unrelated MGUS with these clinical findings.
Tissue biopsy = Gold standard • The diagnosis of cardiac amyloidosis is confirmed either by • demonstrating amyloid deposits on endomyocardial biopsy • or, in patients with appropriate cardiac findings, by demonstrating amyloid deposits on histologic examination of a biopsy from other tissues (eg, abdominal fat pad, rectum, or kidney).
Medication Interaction • Amyloid fibrils bind to both digoxin and nifedipine • Increased susceptibility to digitalis toxicity and to hemodynamic deterioration after nifedipine • Angiotensin converting enzyme (ACE) inhibitors often provoke profound hypotension in AL amyloidosis, possibly by exposing a subclinical autonomic neuropathy. • Amiodarone seems to be relatively well tolerated strategy for rate control in atrial fibrillation.
Treatment Options • Melphan + steroids • Cyclophosphamide + thalidomide • Autologous HCT • Heart Transplant
Prognosis • Untreated: • Median survival • six to nine months in those with heart failure • 1.1 years in those with any sign of cardiac involvement
Hematopoietic Cell Transplant • Melphalan therapy + autologous HCT has had a significant impact on survival in AL amyloidosis without cardiac involvement • Cardiac amyloidosis is associated with increased morbidity and mortality from HCT and reduced post-therapy survival compared to those without clinically apparent cardiac involvement. • The largest reported experience of HCT comes from an eight-year study of 701 consecutive new patients with AL amyloidosis
Hematopoietic Cell Transplant • 312 were eligible for high-dose melphalan and HCT • Cardiac involvement, (137 patients - 43 %), was defined by • septal or posterior wall thickening ≥13 mm on echocardiography • clinical syndrome of heart failure. • The following observations were noted in the patients with cardiac involvement: • At one year, 21% had a cardiac response, defined as • a decrease in intraventricular septal thickness (if initially increased) of ≥2 mm or • a decrease in NYHA functional class of at least one grade without an increase in diuretic dose. • Median survival was 1.6 years compared to 6.4 years in patients without cardiac involvement. • However, some cardiac patients had a prolonged survival, with approximately one-third alive at five years.
Chemotherapy • Regimens: • melphalan + prednisone • cyclophosphamide, thalidomide and dexamethasone • In a report of 46 patients who were not eligible for HCT (32 because of severe cardiac involvement), the administration of up to nine courses of melphalan + prednisone was associated with a hematologic response in 67 % and complete hematologic remission in 33%. • An organ response was noted in 22 pts (48%), including six with a ≥2 mm reduction in interventricular septum thickness that was associated with resolution of heart failure.
Heart transplantation • The majority with cardiac AL amyloidosis have significant noncardiac amyloidosis and are not suitable candidates for heart transplantation. • In one series, only 4 percent had clinically isolated cardiac disease • Early cardiac transplantation did not address the underlying plasma cell dyscrasia, which later progressed in other organs and/or returned in the transplanted heart. • Heart transplantation is followed by high–dose chemotherapy and autologous HCT within a 12-month period. Long-term follow-up data in these patients is not yet available, but several appear to have had an good results
Summary • AL amyloid cardiomyopathy presents with rapidly progressive symptoms of right-sided heart failure • SPEP, serum free light chains • LV thickening/restrictive cardiomyopathy + low-voltage ECG • Characteristic appearance on TTE and cardiac MRI • Tissue biopsy if possible • Poor prognosis, but some treatment response to chemotherapy and HCT
References • Mullens et al. Resolution of cardiac amyloidosis after autologous blood stem cell transplantation. European Heart Journal. • Kyle, RA. "Amyloidosis: The Last Three Centuries." Amyloid and Amyloidosis. Bely, M, Apathy, A (Eds), 2001; p10-13. • Falk and Skinner. The systemic amyloidose: an overview. Adv Intern Med 2000; 45:107. • Maurer et al. Cardiac transplantation using extended-donor criteria organs for systemic amyloidosis complicated by heart failure. Transplantation 2007; 83:539. • Up To Date. Amyloid Cardiomyopathy.