Download

1 / 41
450 likes | 672 Views
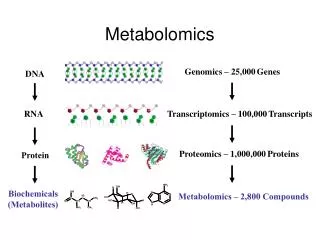
Statistics in Metabolomics. David Banks ISDS Duke University. 1. Background. Metabolomics is the next step after genomics and proteomics. There are about 25,000 genes, most of which have unknown functions. There are about 1,000,000 proteins, most of which are unstudied.

E N D
Statistics in Metabolomics David Banks ISDS Duke University
1. Background Metabolomics is the next step after genomics and proteomics. There are about 25,000 genes, most of which have unknown functions. There are about 1,000,000 proteins, most of which are unstudied.
In contrast to the *omics areas: • There are only about 900 main metabolites, and we know their chemical structures • Also, we know (pretty well) the biochemical pathways that determine their production rates Metabolites are low-weight molecular compounds produced in the course of processing raw materials.
Some common metabolites include: • cholesterol • glucose, sucrose, fructose • amino acids • lactic acid, uric acid • ATP, ADP • drug metabolites, legal and illegal These are produced in metabolic pathways, such as the Krebs (citrate) cycle for oxidation of glucose.
These pathways contain important information about the amount of each metabolite: • Stoichiometric equations show how much material is produced in a given reaction; i.e., mass balance. • Rate equations govern the speed at which reactions take place, and the location of the Gibbs equilibrium This gives metabolomics an edge.
Biochemical Profile Biochemical Profile Map to Metabolic Pathways
The purposes of metabolomics are: • Early detection of disease, such as necrosis, ALS, Alzheimer’s, and infection or inflammation. • Assessment of toxicity (especially liver toxicity) in new drugs. • Diet strategies, drug testing. • Elucidating biochemical pathways. There is less raw information than for other *omics, but more context.
2. Measurement Issues To obtain data, a tissue sample is taken from a patient. Then: • The sample is prepped and put onto wells on a silicon plate. • Each well’s aliquot is subjected to gas and/or liquid chromatography. • After separation, the sample goes to a mass spectrometer.
The sample prep involves stabilizing the sample, adding spiked-in calibrants, and creating multiple aliquots (some are frozen) for QC purposes. This is roboticized. Sources of error in this step include: • within-subject variation • within-tissue variation • contamination by cleaning solvents • calibrant uncertainty • evaporation of volatiles.
Gas chromatography creates an ionized aerosol, and each droplet evaporates to a single ion. This is separated by mass in the column, then ejected to the spectrometer. Sources of error in this step include: • imperfect evaporation • adhesion in the column • ion fragmentation or adductance
The fourier mass spectrometer determines the mass to charge ratio of the ion from the field strength required to keep the ion spinning in a circle. This avoids the entry-time uncertainty in TOF machines, so the only main error is uncertainty about the field strength Some laboratories use MALDI-TOF equipment, and the error sources are slightly different.
The result of this is a set of m/z ratios and timestamps for each ion, which can be viewed as a 2-D histogram in the m/z x time plane. One now estimates the amount of each metabolite. This entails normalization, which also introduces error. The caveats pointed out in Baggerley et al. (Proteomics, 2003) apply.
3. Statistical Problems • Understanding the uncertainty budget in metabolomic data, which entails both quality control and cross-platform comparisons. • Identifying the peaks in the m/z x t plane, and estimating quantity of specific metabolites. • Finding markers for disease or toxicity, or measuring change.
3.1 Uncertainty The classical NIST approach to this is to: • build a model for the error terms • do a designed experiment with replicated measurements • fit a measurement equation to the data See Cameron, “Error Analysis,” ESS Vol. 9, 1982.
Let z be the vector of raw data, and let x be the estimates. Then the measurement equation is: G(z) = x = µ+ ε where µis the vector of unknown true values and εis decomposable into separate components. For metabolite i, the estimate Xiis: gi(z) = lnΣ wij∫∫sm(z) – c(m,t)dm dt.
The law of propagation of error (this is essentially the delta method) says that the variance in X is about Σni=1 (∂g /∂ zi)2 Var[zi] + Σi≠k 2 (∂g/∂zi)(∂g/∂zk) Cov[zi, zk] The weights depend upon the values of the spiked in calibrants, so this gets complicated.
Cross-platform experiments are also crucial for medical use. This leads to key comparison designs. Here the same sample (or aliquots of a standard solution or sample) are sent to multiple labs. Each lab produces its spectrogram. It is impossible to decide which lab is best, but one can estimate how to adjust for interlab differences.
The Mandel bundle-of-lines model is what we suggest for interlaboratory comparisons. This assumes: Xik = αi + βiθk + εik where Xik is the estimate at lab i for metabolite k, θk is the unknown true quantity of metabolite k, and εik ~ N(0,σik2).
To solve the equations given values from the labs, one must impose constraints. A Bayesian can put priors on the laboratory coefficients and the error variance. Metabolomics needs a multivariate version, with models for the rates at which compounds volatilize. We plan to use this model to compare the Metabolon lab in RTP to Chris Newgard’s lab at Duke.
3.2 Peak Identification A classic problem in proteomics is to locate peaks and estimate their area or volume. Unlike proteomics, metabolite peak location is mostly known. So Bayesian methods seem good (cf. Clyde and House). Metabolon uses proprietary software.
3.3 Data Mining Different tools are appropriate for different kinds of metabolomic studies. The work we have done focuses on: • Random Forests • Support Vector Machines • Robust Singular Value Decomposition
We had abundance data on 317 metabolites from 63 subjects. Of these, 32 were healthy, 22 had ALS but were not on medication, and 9 had ALS and were taking medication. The goal was to classify the two ALS groups and the healthy group. Here p>n. Also, some abundances were below detectability.
Using the Breiman-Cutler code for Random Forests, the out-of-bag error rate was 7.94%; 29 of the ALS patients and 29 of the healthy patients were correctly classified. 20 of the 317 metabolites were important in the classification, and three were dominant. RF can detect outliers via proximity scores. There were four such.
Several support vector machine approaches were tried on this data: • Linear SVM • Polynomial SVM • Gaussian SVM • L1 SVM (Bradley and Mangasarian, 1998) • SCAD SVM (Fan and Li, 2000) The SCAD SVM had the best loo error rate, 14.3%.
The L1 SVM attempts to mimic the automatic variable selection in the LASSO (Tibshirani, 1996) by solving the programming problem: Minb,wΣ[1 – yi(b+wTxi)]+ + λΣ | wk | where the first sum is over n and the second is over p. SCAD replaces the L1 penalty with a nonconvex penalty.
The SCAD SVM selected 18 of the metabolites as being important; the L1 selected 32. This suggests that the automatic variable selection in L1 SVM is not very effective. A further multiple tree analysis with FIRMPlusTM software from the GoldenHelix Co. did not achieve good classification. So Random Forests wins. And the selected metabolites make sense.
Robust SVD (Liu et al., 2003) is used to simultaneously cluster patients (rows) and metabolites (columns). Given the patient by metabolite matrix X, one writes Xik = ri ck + εik where ri and ck are row and column effects. Then one can sort the array by the effect magnitudes.
To do a rSVD use alternating L1 regression, without an intercept, to estimate the row and column effects. First fit the row effect as a function of the column effect, and then reverse. Robustness stems from not using OLS. Doing similar work on the residuals gives the second singular value solution.
3.3.1 Preterm Labor The NIH wanted to decide whether amniotic fluid samples from women in preterm labor could support classification: • Term delivery • Preterm delivery with inflammation • Preterm delivery without inflammation.
The analysis had samples from 113 women in preterm labor. We tried all of the usual classification methods. As before, Random Forests gave the best results. The various SVMs were about 5-10% less predictive. The main information was contained in amino acids and carbohydrates.
Predicted Term Inflamm. No Inf. Term 39 1 0 True Inflamm. 7 32 1 No Inf. 2 2 29 RF accuracy was 100/113 = 88.49%.
For those with term delivery, amino acids were low, carbohydrates were high. For those who had preterm delivery without inflammation, both amino acids and carbohydrates were low. For those who had inflammation, the carbohydrates were very low and the amino acids were high.
My collaborators in this research are: • Chris Beecher, Metabolon, Inc. • Adele Cutler, USU • Leanna House, Duke University • Jackie Hughes-Oliver, NCSU • Xiadong Lin, U. of Cincinnati • Susan Simmons, UNC-Wilmington • Young Truong, UNC-Chapel Hill • Stan Young, NISS