Download
1 / 47
480 likes | 685 Views
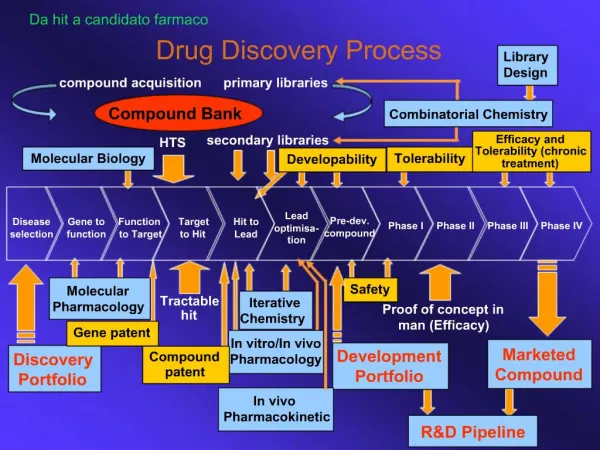
Informationsfluß in einer drug discovery pipeline. eADMET Prediction. early Absorption Distribution Metabolism Elimination Toxicology. Pharmacokinetic Bioavailability. ADME Modelle (I). Folgende Modelle werden für das in silico design benötigt. Primäre Modelle

E N D
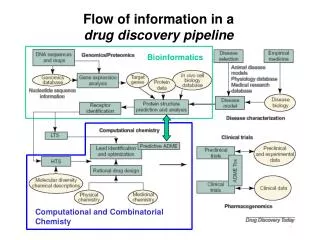
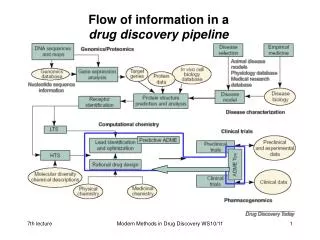
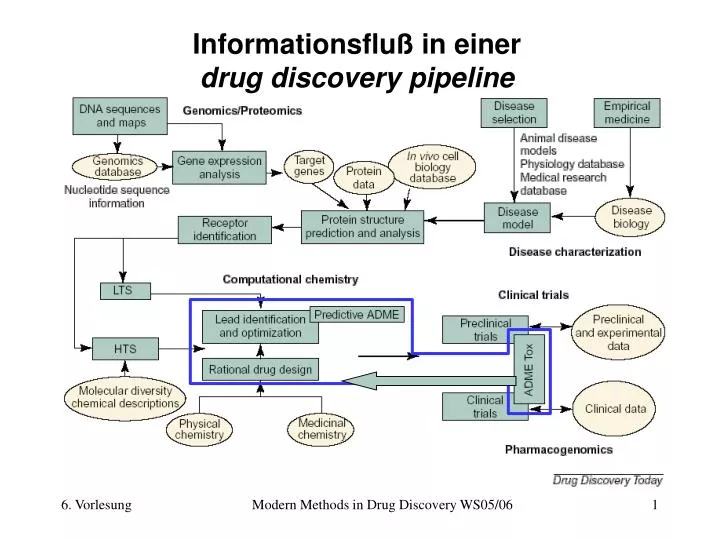
Informationsfluß in einerdrug discovery pipeline Modern Methods in Drug Discovery WS05/06
eADMET Prediction early Absorption Distribution Metabolism Elimination Toxicology Pharmacokinetic Bioavailability Modern Methods in Drug Discovery WS05/06
ADME Modelle (I) Folgende Modelle werden für das in silico design benötigt Primäre Modelle Löslichkeitintestinale AbsorptionBioverfügbarkeitMetabolische StabilitätBlut-Hirn-Schranke PermeationMutagenizitätCardiatische Toxizität (hERG)Plasmaprotein Bindung Sekundäre Modelle Transport (Aufnahme und Efflux)Allgemeine ToxizitätHepatotoxizität (PXR, CAR)NephrotoxizitätImmunogenizitätNeurotoxizität (Rezeptorbindung)Drug-Drug Wechselwirkungen (Cytochrom P450) Modern Methods in Drug Discovery WS05/06
ADME Modelle (II) Modern Methods in Drug Discovery WS05/06
Warum ist die Voraussage der ADME Parameter so wichtig ? Gründe die zum Fehlschlag eines potentiellen Wirkstoffs führen Modern Methods in Drug Discovery WS05/06
Warum ist die Voraussage der ADME Parameter so wichtig ? (II) • Zeil ist es, ungeeignete Verbindungen möglichst frühzeitig zu erkennen: • Schonung von Resourcen • Vermeidung unnötiger klinischer Tests • Je später ein Wirkstoff zurückgezogen werden muß, desto teuerer wird es. • „Fail early, fail fast, fail cheap“ Modern Methods in Drug Discovery WS05/06
Komponentenauswahl für dasHigh Throughput Screening (HTS) Typischer eADME Filter Modern Methods in Drug Discovery WS05/06
Solvatation kontra Löslichkeit DGsolv logS Modern Methods in Drug Discovery WS05/06
Löslichkeitsmodelle (I) • Die Berechnung der Löslichkeit aus einem thermodynamischen Zyklus (Gitterenergie, Solvatationsenergie) wäre prinzipiell möglich, jedoch ist • Die Voraussage der Gitterenergie praktisch kaum möglich da dies die Raumgruppe des Kristalls erfordert • Die Berechnung der Solvatationsenerge selbst fehlerbehaftet ist Deshalb kommen vorwiegend QSAR Ansätze zur Anwendung Modern Methods in Drug Discovery WS05/06
Löslichkeitsmodelle (II) Deskriptoren: Konnektivitätsindices r2=0.89, q2= 0.84, se = 0.98, n=120, F=297.80 Lit. C. Zhong et al. J.Pharm.Sci.92 (2003) 2284 Modern Methods in Drug Discovery WS05/06
Löslichkeitsmodelle (III) Weitere Ansätze zeigen, daß die verwendeten Deskriptoren die Lipophilie, H-Brückenbindungseigenschaften und Flexibilität der Verbindungen wiedergeben müssen Lit: A. Cheng et al. J.Med.Chem.46 (2003) 3572 D. Butina et al. J.Chem.Inf.Comput.Sci.43 (2003) 837 Neben klassischen QSAR-Gleichungen kommen zunehmend auch Neuronale Netze zum Einsatz Lit: A. Yan et al. J.Chem.Inf.Comput.Sci.43 (2003) 429 J.K. Wegener et al. ibid43 (2003) 1077 Modern Methods in Drug Discovery WS05/06
Absorption Wieviel und wie schnell wird der Wirkstoff aufgenommen ? Praktischerweise sollte ein Pharmakon oral appliziert werden können Es wird nach der Magen-passage vom Darm ins Blut resorbiert. (Über die Pfort-ader zunächst in die Leber) Modern Methods in Drug Discovery WS05/06
Absorption im duodenum (I) Aufnahme des Wirkstoffes in den Blutkreislauf Ausschnitt aus der Darmwand Modern Methods in Drug Discovery WS05/06
Absorption im duodenum (II) Aufnahme des Wirkstoffes in den Blutkreislauf Ausschnitt aus der Darmwand Modern Methods in Drug Discovery WS05/06
Absorption im duodenum (III) Modell der Zellmembran Phospholipid De Groot et al. Science294 (2001) 2353 Modern Methods in Drug Discovery WS05/06
Caco-2 Zellen Monolayer Experimenteller Ansatz zur Voraussage der intestinalen Absorption Monolayer aus einer Kultur von Zellen dieeigentlich von einem Dickdarmkrebsstammen. Vorteil: Reproduzierbare Ergebnisse,relativ gute Übereinstimmung mit in vivo Studien Nachteil: Diese Zellen besitzen etwas andere metabolische Eigenschaften als Zellen des Dünndarms (MDR1 Transporterüberexprimiert) Daneben kommen neuerdings auch synthetische Membranen zum Einsatz Modern Methods in Drug Discovery WS05/06
Welche Faktoren bestimmen die passive Diffusion durch Lipiddoppelschichten ? Kleine Moleküle sollten die Membran schneller durchdringen als größere Deskriptor: Molekülgewicht (MW) und Molekülform Phospholipiddoppelschichten sind im Inneren lipophil Deshalb sollten lipophile Moleküle sie schneller passieren Deskriptor: logP (water/n-octanol partition coefficient) Phospholipiddoppelschichten weisen eine hydrophile äußere Oberfläche auf Deskriptoren: Anzahl von H-Brücken Donoren und Akzeptoren Beobachtung: Die Permeation geht mit der Lösungswärme einher Modern Methods in Drug Discovery WS05/06
Molekülbasierte Deskriptoren zurVoraussage der ADME Eigenschaften logP Wasser/Octanol Verteilungskoeffizient Lipinski‘s rule Topologische Indices Polar surface area Similarität / Dissimilarität QSAR quantitative structure activity relationship QSPR quantitative structure property relationship Modern Methods in Drug Discovery WS05/06
Lipinski´s Rule of 5 Kombination von Deskriptoren zur Abschätzung der intestinalen Absorption. Schlechte Aufnahme der Verbindung, wenn Molekülmasse > 500 logP > 5.0 > 5 H-Brücken Donoren (OH und NH) >10 H-Brücken Akzeptoren (N und O) Schlechte Diffusion Zu lipophil Zuviele H-Brücken mit den Kopfgruppen der Membran C.A. Lipinski et al. Adv. Drug. Delivery Reviews23 (1997) 3. Modern Methods in Drug Discovery WS05/06
Polar Surface Area (PSA) Die PSA ist definiert als der Bereich der van der Waals Oberfläche des Moleküls, der von den polaren Stickstoff- und Sauerstoffatomen, sowie den an sie gebundenen Wasserstoffen herrührt. Maß für die Ausbildung von H-Brücken Wie bei allen 3D Deskriptoren ist auch die PSA im Prinzip abhängig von der Konformation. Modern Methods in Drug Discovery WS05/06
Absorptionsmodelle Neue Arbeiten zeigen allerding, daß auch ohne Berücksichtigung verschiedener Konfomere eine gute Korrelation zur Caco-2 Absorption und Aufnahme im Menschen (%FA) besteht. Vollständige Aufnahme (>90%) wenn PSA<60 A2 Schlechte Aufnahme (<10%) wenn PSA>140 A2 Lit: D.E. Clark, J.Pharm.Sci.8 (1999) 807; Drug Discovery Today5 (2000) 49; K. Palm et al. J.Med.Chem.41 (1998) 5382 Modern Methods in Drug Discovery WS05/06
Pharmacokinetik und Bioverfügbarkeit „Quantitative Auseinandersetzung des Organismus mit einem einverleibten Pharmakon“ Der Köper/Organismus wird als offenes System betrachet, daß nach jeder Störung/Arzneimittelzugabe versucht, den Gleich-gewichtszustand wiederherzustellen Dazu teilt man den Körper in eine Reihe von Räumen (compartments) auf, zwischen denen ein Fließgleichgewicht herrscht Modern Methods in Drug Discovery WS05/06
Distribution / Invasion Der Gesamtweg des Medikamentes läßt sich in Einzelschritte aufteilen: • Diffusion im Lösungsmittel • D. durch Gewebs- und Gefäßmembranen • Transport durch das Blut • a) D. zu den target Rezeptoren • b) D. in andere Kompartimente • c) D. in Eliminationsorgane • 5) Irreversible Elimination Absorption Invasion (nach Dost)≈ Distribution Hohe Eliminationskonstante: Kurzzeitnarkotika Niedrige Eliminationskonstante: Antibiotika Modern Methods in Drug Discovery WS05/06
Verteilungsvolumen und Dosierung Die Dosisierung richtet sich nach dem Verteilungsvolumen Modern Methods in Drug Discovery WS05/06
Invasion / System exposure (II) Nur mittels intravenöser Gabe ist die volle Konzentration sofort verhanden. Ansonsten überschneiden sich Invasion und Elimination. Dies entspricht physicochemisch einer Folgereaktion Nur Invasion ▬▬ Nur Elimination ▬▬ Rasche Invasion ▬▬ Rasche Elimination ▬▬ Therapeutische Bandbreite Modern Methods in Drug Discovery WS05/06
Das Dost‘sche Prinzip (I) Wie verhält sich der Konzentrationsverlauf bei unterschiedlichen Dosen ? Zwischen zwei Meßzeiten erhält man die Fläche S (Transit) unter der Kurve durch Integration der Batemanfunktion als Total clearance: Volumen das pro Zeiteinheit geklärt wird korrespondierende Flächen entsprechen dem Verhältnis der Dosen Modern Methods in Drug Discovery WS05/06
Das Dost‘sche Prinzip (II) Die Referenzkurve erhält man durch intravenöse Gabe der Dosis Occupancy = meßbare Konzentration Transit = bereits irreversibel eliminierte Menge Transfer = Occupancy + Transit = absorbierte Menge Availments = noch zur Invasion anstehende Menge Modern Methods in Drug Discovery WS05/06
Daten für Pharmakokinetische Modelle Chemische DatenBiologische Daten Verteilungskoeffizienten Anatomische Abmessungen Metabolische Umsatzraten Blutfluß durch OrganeVmax, Km, Ki Organvolumina Löslichkeit Dampfdruck Atmung Diffusionsgeschwindigkeit Körpergewicht Proteinbindungskonstanten Alter, Geschlecht, Ausmaß körperlicher Aktivität Modern Methods in Drug Discovery WS05/06
Pharmakokinetische Modelle (I) KompartimentmodelleAnnahme: keine metabolische Umsetzung in den Verteilungsräumen Leber Darm Blut Niere Konzentrationverlauf läßt sich über lineare Differentialgleichungen berechnen Modern Methods in Drug Discovery WS05/06
Pharmakokinetische Modelle (II) Blutkreislauf als elektrisches Netzwerk (1930) Per Analogrechner simulierbar (variable Steckverbindungen zwischen den Modulen) Anwendbarkeit: Narkotika (geringe Metabolisierung, lipophil, werden abgeatmet) Modern Methods in Drug Discovery WS05/06
Distribution Je nach Ziel muß der Wirkstoff vom Plasma aus weitere Kompartimente erreichen. Bei Wirkstoffen die auf das zentrale Nervensystem wirken muß die Blut-Hirn-Schranke passiert werden. Umgekehrt sollten andere Wirkstoffe die Barriere nicht überwinden. Auch der aktive Transport von Stoffen kommt in Betracht Modern Methods in Drug Discovery WS05/06
Eiweißbindung / Distribution (I) Die Konzentration von Wirkstoffen kann durch Bindung an Eiweiße verringert werden. Dies gilt sowohl für das Plasma, die extracelluläre und die interstitielle Flüssigkeit. Im Gleichgewicht ist keine Veränderung meßbar, und damit Die Bindung erfolgt gemäß der Langmuir‘sche Absorptionsisotherme (Absorptionswärme unabhängig vom Belegungsgrad) und erfüllt damit das Massenwirkungsgesetz Neben Proteinen können auch Mucopolysaccharide (Binde- und Stützgewebe) absorbieren Modern Methods in Drug Discovery WS05/06
Metabolismus (I) (Bio-)chemische Reaktionen von Xenobiotica im Körper First pass effect: Extensive Umsetzung von vorwiegend lipophilen Molekülen, solchen mit MW>500, oder die eine spezifische Affinität zu bestimmten Transportern haben,bei der ersten Passage durch die Leber Phase I: Oxidation, Reduktion und Hydrolyse Cytochrom P450 Enzyme Phase II: Konjugation mit kleinen Molekülen (z.B. Glutamin) Phase III: Elimination durch Transporter Modern Methods in Drug Discovery WS05/06
Metabolismus (II) Experimentelle (in vitro) Methoden:Leber Mikrosomen vom Menschen, Hepatocyten und rekombinante P450 Isozyme (exprimiert in E. coli) Modern Methods in Drug Discovery WS05/06
Elimination / Exkretion Unter Elimination werden alle Vorgänge zusammengefaßt, die zur Entfernung eines Stoffes aus einem Kompartiment führen. Diese können auch metabolischerArt sein. Lipophile Stoffe können über die Galle, hydrophile Stoffe über den Harn ausgeschieden werden. Allgemein gilt: MW <300 300-500 >500 Galle Galle & Harn Harn Modern Methods in Drug Discovery WS05/06
Elimination / Clearance Überblick der metabolischen Pfade Modern Methods in Drug Discovery WS05/06
Elimination / Clearance (III) Physicochemisch gesehen ist die Eliminierung eines Stoffes ein Zerfallsprozess 1. Ordnung (abhängig von der jeweils vorhandenen Konzentration des Stoffes) Modern Methods in Drug Discovery WS05/06
Was ist die Blut-Hirn Schranke (BBB) ? Querschnitt durch ein Blutgefäß Nach: J.-M. Scheerman in Pharmacogenomics, J.Licinio & Ma-Li Wong (Eds.) Wiley-VCH (2002) pp. 311-335. Modern Methods in Drug Discovery WS05/06
Wozu dient die Blut-Hirn Schranke ? • Die in silico Vorhersage der Permeabilität der Blut-Hirn Schranke (BBB) einer Verbindung im Rahmen der vorklinischen Entwicklung ist besonders wichtig, da • Nur Substanzen die auf das zentrale Nervensystem (CNS) wirken, sollen die Blut-Hirn Schranke effektiv überwinden. • BBB-Screening ist besonders „teuer“ (Tierversuche kaum vermeidbar: Mikrodialyse, Isotopenmarkierung) Modern Methods in Drug Discovery WS05/06
Blood-Brain Barrier (BBB) Als Maß für das Durchdringen der Blut-Hirn Schranke wird der Logarithmus des Verhältnis der Konzentrationen angegeben logBB = log([im Hirn]/[imBlut]) Bereich: -2.00 bis +1.00 Vorwiegend im Blut -1.0 < logBB < 0.3 vorwiegend im Hirn Experimentell ist der logBB Wert nur schwer zugänglich (Isotopenmarkierung, Mikrodialyse) und auch Modelle aus künstlichen Membranen (Endothelial Zellen) sind noch in der Entwicklung Lit. D. E. Clark, J. Pharm. Sci.8 (1999) 815 Modern Methods in Drug Discovery WS05/06
Blood-Brain Barrier (II) Im Gegensatz zur Absorption aus dem Duodenum spielt hier die Polarität der Verbindungen eine große Rolle, die nicht durch die PSA beschrieben werden kann. Bsp: PSA logBB ClogP Polarisierbarkeit(AM1) Benzol 0 -0.69 2.1 13.8 3-Methylpentan 0 2.01 3.7 14.8 Entsprechend läßt sich eine QSRP-Gleichung aufstellen logBB = a PSA + b ClogP + c mit r = 0.887 Lit. D. E. Clark, J.Pharm.Sci.8 (1999) 815 F. Lombardo et al. J.Med.Chem.39 (1996) 4750 Modern Methods in Drug Discovery WS05/06
Bisher benutzte Deskriptoren Folgende Terme korrelieren direkt mit logBB: ● logP ● Polar surface area ● hydrogen-bond donors and acceptors ● size and shape fragment based (MlogP, ClogP,...) contributions from N, O and H atoms numerical count molecular volume and globularity Modern Methods in Drug Discovery WS05/06
Deskriptoren für Größe und Form Bezug zur Molekülform haben: Molekülvolumen, Globularität, Anzahl drehbarer Bindungen Globularität: Verhältnis der Oberfläche (unter der Annahme das Molekül wäre eine Kugel) zur tatsächlichen Oberfläche. Immer < 1 Hauptkomponenten der Molekülgeometrie: Ausdehnung des Moleküls im dreidimensionalen Raum Modern Methods in Drug Discovery WS05/06
Neue Deskriptoren für Größe und Form - Deskriptoren wie etwa die Globularität sind korreliert mit dem Molekülgewicht und der Anzahl der H-Atome + Ersatz durch drei Terme die aus der Molekülgeometrie abgeleitet werden PCGC PCGA PCGB Modern Methods in Drug Discovery WS05/06
BBB-Model mit 12 Deskriptoren Deskriptoren hauptsächlich aus QM Rechnungen: Elektrostatische Oberfläche, Hauptkomponenten der Geometrie,H-Brücken Eigenschaften Lit: M. Hutter J.Comput.-Aided.Mol.Des. 17(2003) 415. Modern Methods in Drug Discovery WS05/06
ADME - Historische Entwicklung • Corwin Hansch QSAR für kleine Datensätze • logP für Toxizität 1980 in vitro Studien ersetzen in vivo Studien 1990 Erste in silico ADME Modelle (Computer) Docking in Proteinstrukturen Homologiemodellierung von Proteinen (CYP P450) 1997 Lipinski‘s Rule of Five zur Absorption 2002 X-Ray Struktur von human CYP2C9 2004 X-Ray Struktur von human CYP3A4 Modern Methods in Drug Discovery WS05/06
Web-basierte online Tools Eine Reihe von Instituten und Firmen haben Server für die Vorhersage von ADME-Eigenschaften. Diese basierend zumeist auf einem Java-Applet mit dem die Moleküle gezeichnet werden können, bieten die Eingabe als SMILES-String oder als eines der vielen Formate für3D-Strukturen an. Eine Zusammenfassung mit Hyperlinks bietet das Virtual Laboratory http://146.107.217.178/online.html Lit. I.V. Tetko, Mini Rev.Med.Chem.8 (2003) 809 Modern Methods in Drug Discovery WS05/06