Download

1 / 83
840 likes | 998 Views
Lecture 25 March 04, 2011 Ionic bonding and crystals. Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy. William A. Goddard, III, wag@wag.caltech.edu 316 Beckman Institute, x3093

E N D
Lecture 25 March 04, 2011 Ionic bonding and crystals Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy William A. Goddard, III, wag@wag.caltech.edu 316 Beckman Institute, x3093 Charles and Mary Ferkel Professor of Chemistry, Materials Science, and Applied Physics, California Institute of Technology Teaching Assistants: Wei-Guang Liu <wgliu@wag.caltech.edu> Caitlin Scott <cescott@caltech.edu>
Experimental data M2 phase – partial occupation at M3, M4, M5, and Te1, Te2 sites Formula: Mo4.31V1.36Te1.81Nb0.33O19.81 Unit cell Occupation M3 M4 M4 M5 M5 M4 M4 M3 O Te2 Te1 All V are VIV, there is no VV DeSanto,.; Buttrey.; Grasselli,.; Lugmair.; Volpe.; Toby.; Vogt, Z. Kristallographie2004, 219, 152-165.
Initial Structure M2 phase To resolve partial occupations • 2x3x4 super cell • 25.26Å x 21.88Å x 16.08Å • 28*24 = 672 atoms • 10 different initial structures • Te : 48 • M3: Mo=13/V=11 • M4: Mo=75/V=21 • M5: Mo=24 [010] [100] 1.95x1027 configurations, Use Monte Carlo
Monte Carlo Swap Simulation 400,000 MC-steps Displacement temperature: 500K Displacement Details: • step-size: 0.1 Å • MC-swap temperature: 25000K • Swap energy bias: 10.0 kcal/mol • Swap frequency: 5 Analysis Method (001) face [001] [010] Each Vertical Column [100]
Conformation of Te-O chain M3 M5 Te1 BVS:3.372 Te2 BVS:4.119 M4 M4 M4 M4 Te1 Te2 No Vanadium M5 M3 M3 M5 M4 M4 M4 M4 M5 M3 Tellurium has zigzag conformation in the center of channel
Distribution of Nb in M5 siteshave 6 M5 sites in 2x3 supercell 8 Nb (open) and 16 Mo (filled) M3 M4 M4 z Nb column M5 M5 1 M4 M4 M3 2 3 4 5 Mo Nb prefer to stay one column Nb
Distribution of V,Mo in M3 and M4 sites when M5 site is Nb (not Mo) Mo Mo Nb Mo Mo Mo Types of Vertical Distribution C Green VVMoMo E Blue VMoMoMo F Purple MoMoMoMo A Red VVVV B Orange VVVMo y x Mo prefers nucleate VVVV May be best for activation of propane Nb decreases the fraction of VVVV May decrease activity for propane But may increase selectivity
QM calculation on Te-O chain, replacing in-plane Te-O-M bonds with Te-OH H2O TeF4 is hypervalent with in-plane bonds at ~95° and out-of-plane bonds at ~180°. For M2 Phase we find Te-O-Te-O-Te-O hypervalent chains
Expect that Te=O can extract allylic Hydrogen H All Te are TeIV before and after the H abstraction Te=O extracts Allylic H to from TeOH while allyl trapped on nearby Mo=NH H2O Continue with previous mechanism for propene on BiMoOx
Surface of M2 Build Process Top and Bottom surface oxygen The final surface contains 71 oxygen atoms(74.0% coverage), 32 on the top and 39 on the bottom (totally 96 oxygen positions)
Active site for propene activation H-allyl bondenergy:-88.0kcal/mol Use H atom to test activity of each site of the surface H added to the surface TeO is the most active site of M2 phase for abstracting Hydrogen from propene TeO—H
Active center M4 M5 M4 M4 M3 M4 M4 M5 M4 vertical oxygen are hidden • Active site can contain one V, two V or three V • one VIV one TeIV exists in the channel • 2V 2Te, 3V 3Te • M5 definitely would contain some NbV if there are only one or two VIV in the center
Conclusion about M2 phase Vanadium prefers to stay as one column in M3 and M4 sites Nb has effect of segregating the Vanadium columns Te-O in the center of channel in zigzag conformation, TeIV Surface coverage of oxygen on M2 surface is 74.0% TeO hypervalent chain is the most active site for abstracting allyic H from propene
Reactions of hydrocarbons on Ni468 nanoparticle Jan. 20, 2010 New paper on ReaxFF 6 cases: 120 methane, 60 ethene, 60 ethyne, 40 propene, 20 benzene, 20 Cylclohexane Initial and Final structures for ReaxFF RD simulation of 40 propene molecules adsorbing and decomposing on a Ni468 cluster Ni 468 particle, 21A diameter
ReaxFF: Acetylene Adsorption & Decomposition on Ni468 nanoparticle Start: 60 C2H2 Conclusions Both C-H bonds break before the C-C bond breaks Formation of subsurface C helps break C-C bonds. end: 52 Cad + 2 C2H3 gas + 2 C2H2ad + C2Had+C2ad
Ethyne detail Reaction of C with 2nd layer Ni very important Build up surface NixCx in first few rows Dynamics of surface Ni plays important role in dissociating C2 Get some carbon into interior
ReaxFF: Benzene Adsorption & Decomposition on Ni Particle H2 C6Hx C2 C6H6ad Simplified sequence C6H6C6H5C6H4C6H3C5H3 C5H2C4H2C4HC3HC3 C2C At the end 7 6 46
Benzene detail Benzene chemisorbs horizontally on the Ni particle surface through pi electrons. As H removed, get strong C-Ni sigma bonds, reorienting benzene vertically. C atoms denuded of H are “swallowed” by the particle by Pac-Man mechanism, for cleaving C-C bonds. C-H bonds far from the surface are protected until the C atoms separating them from the surface are “eaten” away. C6H6 chemisorbed C6H3-allyl chemisorbed C3H with bare C in subsurface C6H3-allyl tail in surface
Early Stages of CNT Growth from Acetylene Feedstock at 1500K on Ni468 nanoparticle (21A) 2 nanosec NVT-RD • NVT-RD • 1 nanosecond + • 1000 K, 1500 K, 2000K, 2500 K 0.5 fs time- step • 100 fs T-damping Start with 100 gas phase C2H2 molecules, add an additional 50 molecules every 200 ps. At end 350 C2H2
CNT Nucleation Study (after 2ns ReaxFF RD) At the end of the simulation we are left with a large carbon ring structure (C367H78)
Subsurface Analysis of Acetylene Feedstock Decomposition on Ni nanoparticle 300 acetylene molecules after 2 ns RD at 2500K Cross section side view C atoms penetrate 10.5A to the core of the catalyst particle forming nickel carbide H penetrates only part way in, preferring the surface. Cross section head on
Hofmann, S. et al. Nano Lett.2007, 7, 602. Experimental Confirmation of a Yarmulke Mechanism Atomic-scale, video-rate environmental transmission microscopy was used to monitor the nucleation and growth of single walled nanotubes.
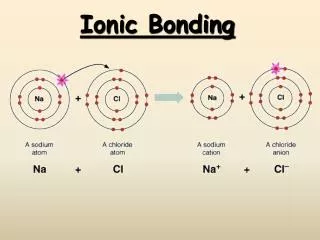
Ionic bonding (chapter 9) Consider the covalent bond of Na to Cl. There Is very little contragradience, leading to an extremely weak bond. Alternatively, consider transferring the charge from Na to Cl to form Na+ and Cl-
The ionic limit At R=∞ the cost of forming Na+ and Cl- is IP(Na) = 5.139 eV minus EA(Cl) = 3.615 eV = 1.524 eV But as R is decreased the electrostatic energy drops as DE(eV) = - 14.4/R(A) or DE (kcal/mol) = -332.06/R(A) Thus this ionic curve crosses the covalent curve at R=14.4/1.524=9.45 A Using the bond distance of NaCl=2.42A leads to a coulomb energy of 6.1eV leading to a bond of 6.1-1.5=4.6 eV The exper De = 4.23 eV Showing that ionic character dominates E(eV) R(A)
GVB orbitals of NaCl Dipole moment = 9.001 Debye Pure ionic 11.34 Debye Thus Dq=0.79 e
electronegativity To provide a measure to estimate polarity in bonds, Linus Pauling developed a scale of electronegativity () where the atom that gains charge is more electronegative and the one that loses is more electropositive He arbitrarily assigned =4 for F, 3.5 for O, 3.0 for N, 2.5 for C, 2.0 for B, 1.5 for Be, and 1.0 for Li and then used various experiments to estimate other cases . Current values are on the next slide Mulliken formulated an alternative scale such that M= (IP+EA)/5.2
Electronegativity Based on M++
Ionic crystals Starting with two NaCl monomer, it is downhill by 2.10 eV (at 0K) for form the dimer Because of repulsion between like charges the bond lengths, increase by 0.26A. A purely electrostatic calculation would have led to a bond energy of 1.68 eV Similarly, two dimers can combine to form a strongly bonded tetramer with a nearly cubic structure Continuing, combining 4x1018 such dimers leads to a grain of salt in which each Na has 6 Cl neighbors and each Cl has 6 Na neighbors
The NaCl or B1 crystal All alkali halides have this structure except CsCl, CsBr, CsI (they have the B2 structure)
The CsCl or B2 crystal There is not yet a good understanding of the fundamental reasons why particular compound prefer particular structures. But for ionic crystals the consideration of ionic radii has proved useful
Ionic radii, main group Fitted to various crystals. Assumes O2- is 1.40A NaCl R=1.02+1.81 = 2.84, exper is 2.84 From R. D. Shannon, Acta Cryst. A32, 751 (1976)
Role of ionic sizes in determining crystal structures Assume that the anions are large and packed so that they contact, so that 2RA < L, where L is the distance between anions Assume that the anion and cation are in contact. Calculate the smallest cation consistent with 2RA < L. RA+RC = (√3)L/2 > (√3) RA Thus RC/RA > 0.732 RA+RC = L/√2 > √2 RA Thus RC/RA > 0.414 Thus for 0.414 < (RC/RA ) < 0.732 we expect B1 For (RC/RA ) > 0.732 either is ok. For (RC/RA ) < 0.414 must be some other structure
Radius Ratios of Alkali Halides and Noble metal halices Rules work ok B1: 0.35 to 1.26 B2: 0.76 to 0.92 Based on R. W. G. Wyckoff, Crystal Structures, 2nd edition. Volume 1 (1963)
Radius rations B3, B4 The height of the tetrahedron is (2/3)√3 a where a is the side of the circumscribed cube The midpoint of the tetrahedron (also the midpoint of the cube) is (1/2)√3 a from the vertex. Hence (RC + RA)/L = (½) √3 a / √2 a = √(3/8) = 0.612 Thus 2RA < L = √(8/3) (RC + RA) = 1.633 (RC + RA) Thus 1.225 RA < (RC + RA) or RC/RA > 0.225 Thus B3,B4 should be the stable structures for 0.225 < (RC/RA) < 0. 414
Structures for II-VI compounds B3 for 0.20 < (RC/RA) < 0.55 B1 for 0.36 < (RC/RA) < 0.96
CaF2 or fluorite structure Like GaAs but now have F at all tetrahedral sites Or like CsCl but with half the Cs missing Find for RC/RA > 0.71
Rutile (TiO2) or Cassiterite (SnO2) structure Related to NaCl with half the cations missing Find for RC/RA < 0.67
CaF2 rutile CaF2 rutile
Electrostatic Balance Postulate For an ionic crystal the charges transferred from all cations must add up to the extra charges on all the anions. We can do this bond by bond, but in many systems the environments of the anions are all the same as are the environments of the cations. In this case the bond polarity (S) of each cation-anion pair is the same and we write S = zC/nC where zC is the net charge on the cation and nC is the coordination number Then zA = Si SI = Si zCi /ni Example1 : SiO2. in most phases each Si is in a tetrahedron of O2- leading to S=4/4=1. Thus each O2- must have just two Si neighbors
a-quartz structure of SiO2 Each Si bonds to 4 O, OSiO = 109.5° each O bonds to 2 Si Si-O-Si = 155.x ° Helical chains single crystals optically active; α-quartz converts to β-quartz at 573 °C rhombohedral (trigonal)hP9, P3121 No.152[10] From wikipedia
Example 2 of electrostatic balance: stishovite phase of SiO2 The stishovite phase of SiO2 has six coordinate Si, S=2/3. Thus each O must have 3 Si neighbors Rutile-like structure, with 6-coordinate Si; high pressure form densest of the SiO2 polymorphs tetragonaltP6, P42/mnm, No.136[17] From wikipedia
TiO2, example 3 electrostatic balance Example 3: the rutile, anatase, and brookite phases of TiO2 all have octahedral Ti. Thus S= 2/3 and each O must be coordinated to 3 Ti. top anatase phase TiO2 right front
Corundum (a-Al2O3). Example 4 electrostatic balance Each Al3+ is in a distorted octahedron, leading to S=1/2. Thus each O2- must be coordinated to 4 Al