Download

1 / 24
270 likes | 571 Views
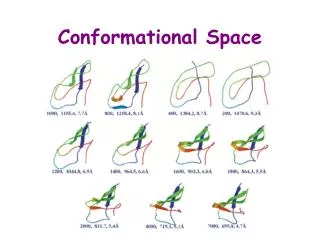
Protein conformational disorders Amyloid. Alice Skoumalová. Hypothetical protein folding pathway: (hierarchical) local segments of secondary structure tertiary structure (subdomains, domains) stable conformation. Local minimum (alternative conformation). Global minimum (native state).

E N D
Protein conformational disordersAmyloid Alice Skoumalová
Hypothetical protein folding pathway: • (hierarchical) • local segments of secondary structure • tertiary structure (subdomains, domains) • stable conformation
Local minimum (alternative conformation) Global minimum (native state) • the protein folding proceeds from a disordered state to progressively more ordered conformations corresponding to lower energy levels • there are more ways of folding (the same protein can aquire more conformations; alternative conformations are represented by local energy minima) Alternative conformations: various function of the protein disease-associated protein
α-helix β-sheet Conformational change • the starting point is the natural protein folded in the native and active conformation • normal protein is rich in α-helix conformations (folded structure) • the end-point is the same protein adopting prevalent β-sheets structure • it is disease-associated protein (misfolded structure) Aggregation Gain of toxic activity Loss of biological function
The conformational change • a change in the secundary or tertiary structure of a normal protein without alteration of the primary structure • the biological function of a protein depends on its tridimensional structure Protein conformatinal disorders (PCD) • diverse diseases arise from protein misfolding • the conformational change may promote the disease by either gain of a toxic activity or by the lack of biological function of the natively folded protein
Protein misfolding causes disease! • the hallmark event in PCD is a formation of β-sheet conformations • the production of β-sheets is usually stabilized by protein oligomerization and aggregation • the misfolded protein self-associates and becomes deposited in amyloid-like aggregates in diverse organs, inducing tissue damage and organ dysfunction
Three different hypotheses have been proposed to describe the relationship between conformational changes and aggregation Polymerization hypothesis Aggregation induces the protein conformational changes Conformational hypothesis Protein misfolding is independent of aggregation, which is a non-necessary end point of conformational changes (the factors inducing the protein structural changes are e.g. mutations, oxidative stress)
Conformation-oligomerization hypothesis Slight conformational changes result in the formation of an unstable intermediate which is stabilized by intermolecular interactions with other molecules forming small β-sheet oligomers
Proteins that are not able to achieve the native state: Recognition Degradation(protein quality control system) 1.Chaperones 2. Ubiquitin proteasome system
DNA Ubiquitin Ribosome RNA ATP Chaperones Native protein Misfolded protein Aggregate/fibrillar amyloid Chaperones Proteasome Accumulation (Amyloidoses) Degraded protein Gain of toxicity (Alzheimer disease) Loss of protein function (Cystic fibrosis)
Implication of protein misfolding 1. Gain of toxicity The harmfull effect of the misfolded protein may be due to deleterious gain of function as seen in many neurodegenerative disorders (Alzheimer disease, Parkinson disease, Hungtington disease), in which protein misfolding results in the formation of harmfull amyloid. Neurodegenerative diseases are characterized by the accumulation of misfolded proteins and formation of aggregates 2. Loss of function Other effect of the misfolded protein may be due to loss of function, as observed in cystic fibrosis. There is a mutation in the CFTR sequence 3. Accumulation Protein aggregates are sometimes converted to a fibrillar structure. Fibrils themselves are not toxic but insoluble. Their accumulation cause tissue damage (amyloidoses)
Chaperones • assist other proteins to achieve a functionally active 3D structure • prevent the formation of a misfolded or aggregated structure Molecular chaperones recognise misfolded protein, bind to the hydrophobic surfaces and inhibit aggregation. Most of these molecules are heat shock proteins (formed during thermal damage)-protect against denaturation. Pharmacological chaperones bind to specific conformations and stabilize them. They are effective in rescuing proteins from proteasomal degradation.
Molecular chaperones Hsp 70 - prevent folding of nascent chain Chaperonins – reverse misfolded structures
Therapy Considering that protein misfolding and aggregation are central in the pathogenesis of PCD, a therapy directed to the cause of the disease should aim to inhibit and reverse the conformational changes Development of novel peptides which can destabilize the abnormal conformation might be useful to correct protein misfolding. Misfolded protein is rich in β-sheet sructure, designed peptides prevent and reverse β-sheet formation (β-sheet breakers) Molecular chaperones play an important role in protein folding, chemical and pharmacological chaperones are experimentally studied
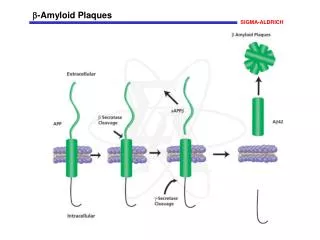
Amyloid • Amyloid is an aggregated protein sructure consisting of unbranched microscopic fibrils often found in dense tissue deposits and associated with a variety of human diseases • The term amyloid does not pertain to a specific protein molecule or sequence, but rather to a general folding motif that appears in various proteins • The amyloid structure exhibit a characteristic folding pattern, called a „cross- β“ structure • Amyloid is a pathogenic structure, formed by accident under conditions of molecular, cellular, or organismic stress, from proteins that evolved to fold and function in different structural states
Polypeptide Major disease states Transthyretin Heart, kidney, peripheral neuropathy Serum amyloid A Kidney, peripheral neuropathy Immunoglobulin light chain Kidney, heart Immunoglobulin heavy chain Spleen β2-Microglobulin Carpal tunnel syndrome, osteoarthropathies Islet amyloid polypeptide Diabetic pancreatic islet cells Fibrinogen α-chain Kidney Apolipoprotein A1 Peripheral neuropathy, liver Atrial natriuretic peptide Heart Amyloid β-protein (Aβ) Brain (Alzheimer‘s disease, cerebral amyloid angiopathy) α-Synuclein Brain (Parkinson‘s disease) Huntingtin polyglutamine Brain (Huntington‘s disease) sequence Prion protein (PrP) Brain (Creutzfeldt-Jakob disease, mad cow disease) Cystatin C, Gelsolin Brain (cerebral amyloid angiopathy) ABri Brain (familial British dementia)
Molecular factors in amyloid formation Protein misfolding is central to amyloid formation Protein stability- the resistance of the folded conformation to misfolding- is an important factor in determining susceptibility to amyloid formation Destabilizing factors: 1. Extreme environments in the body, such as acidic cell compartments 2. Proteolytic removal of a portion of a protein by an endogenous protease 3. Mutations that alter the primary structure (many of the amyloid diseases involve amino acid substitutions in an amyloid precursor protein)
Amyloid fibril structure Straight, unbranched, diameters in the range of 80-160A Composed of two to six protofilaments of diameter 30-40A Rich in a type of β-sheet structure (the β-sheets are perpendicular to the fibril axis and bind together by the hydrogen bonds) β2-microglobulin amyloid fibrils
Overview of amyloid diseases (amyloidosis) Systemic amyloidosis 1. Primary The cause is unknown; abnormal production of immunoglobulins; insoluble protein fibers are deposited in tissues and organs, impairing their function The organs affected: tongue, intestines, skeletal and smooth muscles, nerves, skin, ligaments, heart, liver, spleen, and kidneys 2. Secondary Caused by infection, inflammatory diseases, and sometimes cancer 3.Familial Mutations that make the proteins more prone to aggregation and amyloid deposition (e.g. transthyretin) Organ-specific amyloidosis Diabetes mellitus type 2 (amylin) Alzheimer‘s disease (Aβ) Parkinson‘s disease (α-synuclein) Huntington‘s disease (huntingtin) Transmissible spongioform encephalopathies (prion protein) Cardiac amyloidosis (PrP or transthyretin)
Toxicity of amyloid fibrils 1. Amyloid can cause life-threatening disease by accumulating in such high mass that normal tissue structure and function are disrupted(systemic amyloidosis) 2. The accumulated mass of amyloid is very low compared to the surrounding cell mass (neurodegenerative diseases) 1. Collateral damage caused by immune responses to an amyloid deposits 2. Membrane depolarization resulting from channels created by amyloid fibril assembly intermediates inserted into membranes 3. Recruitment of other proteins into growing aggregates, which has the effect of denying the cell activity of the recruited proteins 4. Disruption of the normal cellular apparatus for breakdown and elimination of misfolded proteins, such as the ubiquitin-proteasome system and the molecular chaperones
Questions • Describe the protein folding funnel • The hallmark event in PCD and consequences • The fate of a misfolded protein in the cell • The role of chaperons • Amyloid - formation, toxicity
Pictures used in the presentation: Marks´ Basic Medical Biochemistry, A Clinical Approach, third edition, 2009 (M. Lieberman, A.D. Marks) Principles of Biochemistry, Third Edition, 2008 (D.J. Voet, J.G. Voet, C.W. Pratt)