Download
1 / 17
550 likes | 2.02k Views
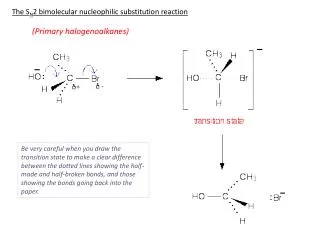
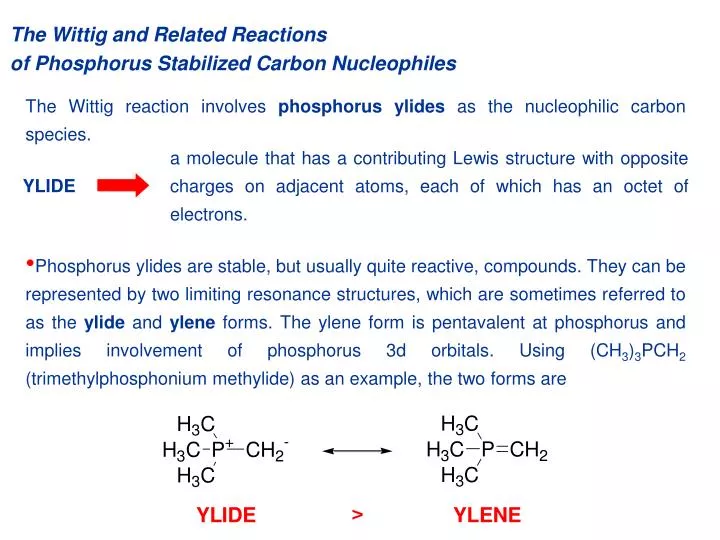
a molecule that has a contributing Lewis structure with opposite charges on adjacent atoms, each of which has an octet of electrons. YLIDE. The Wittig and Related Reactions of Phosphorus Stabilized Carbon Nucleophiles.

E N D
a molecule that has a contributing Lewis structure with opposite charges on adjacent atoms, each of which has an octet of electrons. YLIDE The Wittig and Related Reactions of Phosphorus Stabilized Carbon Nucleophiles The Wittig reaction involves phosphorus ylides as the nucleophilic carbon species. • Phosphorus ylides are stable, but usually quite reactive, compounds. They can be represented by two limiting resonance structures, which are sometimes referred to as the ylide and ylene forms. The ylene form is pentavalent at phosphorus and implies involvement of phosphorus 3d orbitals. Using (CH3)3PCH2 (trimethylphosphonium methylide) as an example, the two forms are
The synthetic potential of phosphorus ylides was initially developed by G. Wittig and his associates at the University of Heidelberg. The reaction of a phosphorus ylide with an aldehyde or ketone introduces a carbon-carbon double bond in place of the carbonyl bond: The mechanism proposed is an addition of the nucleophilic ylide carbon to the carbonyl group to yield a dipolar intermediate (a betaine), followed by elimination of a phosphine oxide. The elimination is presumed to occur after formation of a four-membered oxaphosphetane intermediate. An alternative mechanism might involve direct formation of the oxaphosphetane.
There have been several theoretical studies of these intermediate. Oxaphosphetane intermediates have been observed by NMR studies at low temperature. Betaine intermediates have been observed only under special conditions that retard the cyclization and elimination steps.
Phosphorus ylides are usually prepared by deprotonation of phosphonium salts. The phosphonium salts most often used are alkyltriphenylphosphonium halides, which can be prepared by the reaction of triphenylphosphine and an alkyl halide: • The alkyl halide must be one that is reactive toward SN2 displacement. • Alkyltriphenylphosphonium halides are only weakly acidic, and strong bases must be used for deprotonation. These include organolithium reagents, the sodium salt of dimethyl sulfoxide, amide ion, or substituted amide anions such as hexamethyldisilylamide (HMDS). • The ylides are not normally isolated so the reaction is carried out either with the carbonyl compound present or it may be added immediately after ylide formation. Ylides with nonpolar substituents, for example, H, alkyl, or aryl, are quite reactive toward both ketones and aldehydes.
on the structure of the ylide on the reaction conditions depends strongly stereoselectivity of the Wittig reaction Z-ALKENE UNSTABILIZED YLIDES E-ALKENE STABILIZED YLIDES • Use of sodium amide or sodium hexamethyldisilylamide as bases gives higher selectivity for Z-alkenes than is obtained when ylides are prepared with alkyllithium reagents as base. • The dependence of the stereoselectivity on the nature of the base is attributed to complexes involving the lithium halide salt which is present when alkyllithium reagents are used as bases.
The stereoselectivity of the Wittig reaction is believed to be the result of steric effects which develop as the ylide and carbonyl compound approach one another. • The three phenyl substituents on phosphorus impose large steric demands which govern the formation of the diastereomeric adduct. • Reactions of unstabilized phosphoranes are believed to proceed through an early transition state, and steric factors usually make such transition states selective for the Z-alkene.
A usefull extension of this method is one in which the b-oxido ylide intermediate, instead of being protonated, is allowed to react with formaldehyde. The b-oxido ylide and formaldehyde react to give, on warming, an allylic alcohol. The reaction is valuable for the stereoselective synthesis of Z-allylic alcohols from aldehydes.
The reaction of unstabilized ylides with aldehydes can be induced to yield E-alkenes with high stereoselectivity by a procedure known as the Schlosser modification of the Wittig reaction. • In this procedure, the ylide is generated as a lithium halide complex and allowed to react with an aldehyde at low temperature, presumably forming a mixture of diastereomeric betaine-lithium halide complexes. At the temperature at which the addition is carried out, fragmentation to an alkene and triphenylphosphine oxide does not occur. • This complex is then treated with an equivalent of strong base such as phenyllithium to form a b-oxido ylide. • Addition of t-butyl alcohol protonates the b-oxido ylide stereoselectively to give the more stable syn-betaine as a lithium halide complex. • Warming the solution causes the syn-betaine-lithium halide complex to give the E-alkene by a syn elimination.
The Wittig reaction can be extended to functionalized ylides. Methoxymethylene and phenoxymethylene ylides lead to vinyl ethers, which can be hydrolyzed to aldehydes. • 2-(1,3-Dioxolanyl)methyl ylides can be used for the introduction of a,b-unsaturated aldehydes. Methyl ketones have been prepared by an analogous reaction.
An important complement to the Wittig reaction is the reaction of phosphonate carbanions with carbonyl compounds. • The alkylphosphonate esters are made by the reaction of an alkyl halide, preferably primary, with a phosphite ester. • Phosphonate carbanions are more nucleophilic than an analogous ylide, and even when R is a carbanion-stabilizing substituent, they react readily with aldehydes and ketones to give alkenes. • Phosphonate carbanions are generated by treating alkylphosphonate esters with bases such as sodium hydride, n-butyllithium, or sodium ethoxide. Alumina coated with KF or KOH has also found use as the base.
Reactions with phosphonoacetate esters are used frequently to prepare a,b-unsaturated esters. This is known as the Wadsworth-Emmons reaction. These reactions usually lead to the E-isomer. Three modified phosphonoacetate esters have been found to show selectivity for the Z-enoate product. Trifluoroethyl, phenyl and 2,6-difluorophenyl esters give good Z-stereoselectivity.
An alternative procedure for effecting the condensation of phophonates is to carry out the reaction in the presence of lithium chloride and an amine such as N,N-diisopropyl-N-ethylamine or diazabicycloundecene (DBU). The lithium chelate of the substituted phosphonate is sufficiently acidic to be deprotonated by the amine. Intramolecular reactions have been used to prepare cyclocloalkenes. Intramolecular condensation of phosphonate carbanions with carbonyl groups carried out under conditions of high dilution has been utilized in macrocycle synthesis.
Carbanions derived from phosphine oxides also add to carbonyl compounds. The adducts are stable but undergo elimination to form alkenes on heating with a base such as sodium hydride. This reaction is known as the Horner-Wittig reaction. • The unique feature of the Horner-Wittig reaction is that the addition intermediate can be isolated and purified. This provides a means to control the stereochemistry of the reaction. It is possible to separate the two diastereomeric adducts in order to prepare the pure alkenes. • The elimination process is syn so that the stereochemistry of the alkene depends on the stereochemistry of the adduct. Usually, the anti adduct is the major product, so it is the Z-alkene which is favored. The syn adduct is most easily obtained by reduction of b-keto phosphine oxides.
SUMMARY • Wittig reaction • synthesys of Z allylic alcohols • Schlosser modification • Wadsworth-Emmons reaction • Horner-Wittig reaction
Reactions of Carbonyl Compounds with a-Trimethylsilylcarbanions b-Hydroxyalkyltrimethylsilanes are converted to alkenes in either acidic or basic solution. These eliminations provide a synthesis of alkenes that begins with the nucleophilic addition of an a-trimethylsilyl-substituted carbanion to an aldehyde or ketone. The reaction is sometimes called the Peterson reaction. • For example, the organometallic reagents derived from chloromethyltrimethylsilane adds to an aldehyde or ketone, and the intermediate can be converted to a terminal alkene by base.
Similarly, organolithium reagents of the type (CH3)3SiCH(Li)X, where X is a carbanion stabilizing substituent, can be prepared by deprotonation of (CH3)3SiCH2X with n-butyllithium. • These reagents usually react with aldehydes and ketones to give substitutedalkenes directly. No separate elimination step is necessary because fragmentation of the intermediate occurs spontaneously under the reaction conditions. • In general, the elimination reactions are anti under acidic conditions and syn under basic conditions. • This stereoselectivity is the result of a cyclic elimination mechanism under basic conditions, whereas under acidic conditions an acyclic b-elimination occurs.
The anti elimination can also be achieved by converting the b-silyl alcohols to trifluoroacetate esters. Because the overall stereoselectivity of the Peterson olefination depends on the generation of pure syn or anti b-silyl alcohols, several strategies have been developed for their stereoselective preparation.