Download

1 / 28
290 likes | 484 Views
Gene Finding. Charles Yan. Gene Finding. Genomes of many organisms have been sequenced. We need to translate the raw sequences into knowledge. Where are the genes? How the genes are regulated?. Genome. Human Genome Project (HGP).

E N D
Gene Finding Charles Yan
Gene Finding • Genomes of many organisms have been sequenced. • We need to translate the raw sequences into knowledge. • Where are the genes? • How the genes are regulated?
Human Genome Project (HGP) • To determine the sequences of the 3 billion bases that make up human DNA • 99% human DNA sequence finished to 99.99% accuracy (April 2003) • To identify the approximate 100,000 genes in human DNA (The estimates has been changed to 20,000-25,000 by Oct 2004) • 15,000 full-length human genes identified (March 2003) • To store this information in databases • To develop tools for data analysis
Model Organisms • Finished genome sequences of E. coli, S. cerevisiae, C. elegans, D. melanogaster (April 2003)
Gene Finding • More than 60 eukaryoticgenome sequencing projects are underway
Gene Finding • Thereis still a real need for accurate and fast tools to analyze thesesequences and, especially, to find genes and determine theirfunctions.
Gene Finding • Homology methods, also called `extrinsicmethods‘ • it seems that only approximately half of the genes can be found by homology to other known genes (although this percentage is of course increasing as more genomes get sequenced). • Gene prediction methods or`intrinsic methods‘ • (http://www.nslij-genetics.org/gene/)
Gene Finding • Eukaryotes and Prokaryotes
Gene Finding • Prokaryotes • No introns • The intergenic regions aresmall • Genes may often overlap each other • Thetranslation starts are difficult to predict correctly
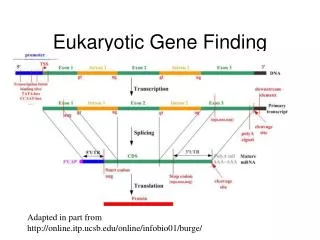
Genes • Functionally, a eukaryotic gene can be defined as beingcomposed of a transcribed region (coding region) and of regions (regulatory region) that cis-regulatethe gene expression, such as the promoter regionwhich controls both the site and the extent of transcription. • The currentlyexisting gene prediction software look only for the transcribedregion (coding region) of genes, which is then called `the gene'.
Genes A gene is furtherdivided into exons and introns, the latter being removed duringthe splicing mechanism that leads to the mature mRNA.
Functional sites (Signals) In the mature mRNA, the untranslated terminalregions (UTRs) are the non-coding transcribed regions,which are located upstream of the translation initiation (5’-UTR) and downstream (3’-UTR) of the translation stop. Theyare known to play a role in the post-transcriptional regulationof gene expression, such as the regulation of translation andthe control of mRNA decay
Functional sites (Signals) Inside or at the boundaries ofthe various genomic regions, specific functional sites (orsignals) are documented to be involved in the various levels ofprotein encoding gene expression. • Transcription (transcriptionfactor binding sites and TATA boxes) • Splicing(donor and acceptor sites and branch points) • Polyadenylation[poly(A) site], • Translation (initiation site, generally ATG withexceptions, and stop codons)
Gene Finding Two different types of information are currentlyused to try to locate genes in a genomic sequence. • (i) Contentsensors are measures that try to classify a DNA region intotypes, e.g. coding versus non-coding. • (ii) Signal sensors aremeasures that try to detect the presence of the functional sitesspecific to a gene.
Gene Finding Content Sensors • Extrinsic content sensors • Base on similarity searching • Intrinsic content sensors • Prediction methods
Extrinsic Content Sensors Extrinsic content sensors The basic tools for detecting sufficient similaritybetween sequences are local alignment methods ranging fromthe optimal Smith-Waterman algorithm to fast heuristicapproaches such as FASTA and BLAST
Extrinsic Content Sensors Similarities with three different types of sequencesmay provide information about exon/intron locations.
Extrinsic Content Sensors The firstand most widely used are protein sequences that can be foundin databases such as SwissProt or PIR. • Pos: Almost 50% of the genes can be identified thanks to a sufficientsimilarity score with a homologous protein sequence. • Neg: Even when a good hit is obtained, a completeexact identification of the gene structure can still remaindifficult because homologous proteins may not share all oftheir domains. • Neg: UTRs cannot be delimited in thisway
Extrinsic Content Sensors The second type of sequences are transcripts, sequenced ascDNAs (a cDNA is a DNA copy of a mRNA) either in theclassical way for targeted individual genes with high coveragesequencing of the complete clone or as expressed sequencetags (ESTs), which are one shot sequences from a wholecDNA library. • Pos: ESTs and `classical' cDNAs are the mostrelevant information to establish the structure of a gene.
Extrinsic Content Sensors Finally, under the assumption that coding sequences aremore conserved than non-coding ones, similarity withgenomic DNA can also be a valuable source of informationon exon/intron location. • Intra-genomiccomparisons can provide data for multigenicfamilies, apparently representing a large percentage of theexisting genes (e.g. 80% for Arabidopsis) (Paralogous genes) • Inter-genomic(cross-species) comparisons can allow the identification oforthologous genes, even without any preliminary knowledgeof them.
Extrinsic Content Sensors • Orthologous:Homologous sequences in different species that arose from a common ancestral gene during speciation. • Paralogous: Homologous sequences in the same speciescaused by a gene duplication occurred in an ancestral species, leaving two copies in all descendants.
Extrinsic Content Sensors Disadvantages of genomic comparisons • Distantly related: The similarity may not cover entirecoding exons but be limited to the most conserved part ofthem. • Closely related: It may sometimes extend to introns and/orto the UTRs and promoter elements. In both cases, exactly discriminatingbetween coding and non-coding sequences is not anobvious task.
Extrinsic Content Sensors Advantages of Extrinsic Content Sensors • An important strength of similarity-basedapproaches is that predictions rely on accumulated preexistingbiological data (with the caveat mentioned later ofpossible poor database quality). They should thus producebiologically relevant predictions (even if only partial). • Another important point is that a single match is enough todetect the presence of a gene
Extrinsic Content Sensors Disadvantages of Extrinsic Content Sensors • Databases may contain information of poorquality • Nothing will befound if the database does not contain a sufficiently similarsequence • Even when a good similarity is found,the limits of the regions of similarity, which should indicateexons, are not always very precise and do not enable anaccurate identification of the structure of the gene. • Smallexons areeasily missed.
Gene Finding Content sensors • Extrinsic content sensors • Compare with protein sequences • Compare with cDNA and ESTs • Genomic comparisons • Intrinsic content sensors • Prediction methods Signal sensors