Download

1 / 27
290 likes | 514 Views
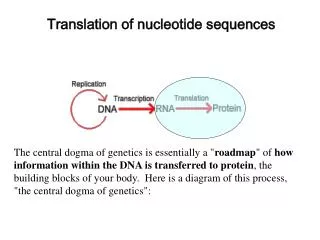
Evolutionary change in nucleotide sequences. Xuhua Xia xxia@uottawa.ca http://dambe.bio.uottawa.ca. Nucleotide substitution. - usually too slow to monitor directly…. spontaneous mutation rates? p. 35-37. for mammalian nuclear DNA (regions not under functional constraint).

E N D
Evolutionary change in nucleotide sequences Xuhua Xia xxia@uottawa.ca http://dambe.bio.uottawa.ca
Nucleotide substitution - usually too slow to monitor directly… spontaneous mutation rates? p. 35-37 for mammalian nuclear DNA (regions not under functional constraint) ~ 4 x 10 -9 nt sub per site per year ... much higher for viruses eg. 10 -6 to 10 -3 nt sub per site per generation … so use comparative analysis of 2 sequences which share a common ancestor • determine number and nature of nt substitutions that have • occurred (iemeasure degree of divergence)
Potential pitfalls 1. Are all evolutionary changes being monitored? - if closely-related, high probability only one change at any given site… but if distant, may have been multiple substitutions (“hits”) at a site - can use algorithms to correct for this 2. If indels between two sequences, can they be aligned with confidence? - algorithms with gap penalties
Ancestral sequence 13 substitutions occurred but only 3 discovered by sequence comparison Present day sequences Fig. 3.6
Multiple hits Homoplasy: same nt, but not directly inherited from ancestral sequence (If comparing long stretches, highly unlikely they would have converged to the same sequence) Page & Holmes Fig. 5.9
Uncertainty in codon substitution Pathway I: CCC(Pro)CCA(Pro) CAA(Gln) Pathway II: CCC(Pro)CAC(His) CAA(Gln) Is one pathway more likely than another? p.82
The purpose of sequence alignment • Identification of sequence homology and homologous sites • Homology: similarity that is the result of inheritance from a common ancestor (identification and analysis of homologies is central to phylogenetics). • An Alignment is an hypothesis of positional homology between bases/Amino Acids.
Normal and Thalassemia HBb 10 20 30 40 50 60 ----|----|----|----|----|----|----|----|----|----|----|----|-- Normal AUGGUGCACCUGACUCCUGAGGAGAAGUCUGCCGUUACUGCCCUGUGGGGCAAGGUGAACGU Thalass. AUGGUGCACCUGACUCCUGAGGAGAAGUCUGCCGUUACUGCCCUGUGGGGCAAGGUGAACGU ************************************************************** 70 80 90 100 110 120 --|----|----|----|----|----|----|----|----|----|----|----|---- Normal GGAUGAAGUUGGUGGU-GAGGCCCUGGGCAGGUUGGUAUCAAGGUUACAAGACAGG...... Thalass. GGAUGAAGUUGGUGGUUGAGGCCCUGGGCAGGUUGGUAUCAAGGUUACAAGACAGG...... **************** *************************************** • Are the two genes homologous? • What evolutionary change can you infer from the alignment? • What is the consequence of the evolutionary change?
What is an optimal alignment? • Alignment1: Favorite- Favourite • Alignment2: ---Favorite Favourite-- • Alignment3: Favorite------ -----Favourite • Alignment4: Favo-rite Favourite • An optimal alignment • One with maximum number of matches and minimum number of mismatches and gaps • Operational definition: one with highest alignment score given a particular scoring scheme (e.g., match: 2, mismatch: -1, gap: -2) • Which of the 4 alignments above is the optimal alignment? • Changing the scoring scheme may change the optimal alignment
Importance of scoring schemes • Two alternative alignments:Alignment 1: ACCCAGGGCTTA ACCCGGGCTTAGAlignment 2: ACCCAGGGCTTA- ACCC-GGGCTTAG • Scoring scheme 1: Match: 2, mismatch: 0, gap: -5 • Scoring scheme 2: Match: 2, mismatch: -1, gap: -1 • Which of the two is the optimal alignment according to scoring scheme 1? Which according to scoring scheme 2? • Importance of biological input
Dynamic Programming Constant gap penalty: Scoring scheme: Match (M): 2 Mismatch (MM): -1 Gap (G): -2 For each cell, compute three values: Upleft value + IF(Match, M, MM)Left value + GUp value + G
Alignment with secondary structure Sequences: Seq1: CACGACCAATCTCGTGSeq2: CACGGCCAATCCGTG Seq1: CACGA ||||| GUGCU Seq2: CACGA ||||| GUGCU Seq2: CACGG ||||| GUGCC CCAAUC Deletion CCAAU Missing link CCAAU Correlated substitution Conventional alignment: Seq1: CACGACCAATCTCGTG Seq2: CACGGCCAATC-CGTG Correct alignment: Seq1: CACGACCAATCTCGTG Seq2: CACGGCCAAT-CCGTG Hickson et al., 2000; Kjer, 1995; Notredame et al., 1997
Multiple Alignment: Guide Tree Seq16 Seq15 Seq14 Seq13 Seq12 Seq11 Seq10 Seq9 Seq8 Seq7 Seq6 Seq5 Seq4 Seq3 Seq2 Seq1 ATTCCAAG... ATTCCAAG... ATTTCCAAG... ATTCCCAAG... ATCGGAAG... ATCCGAAG... ATCCAAAG... ATCCAAAG... AATTCCAAG... AATTCCAAG... AATTTCCAAG... AATTCCCAAG... AAGTCCAAG... AAGTCCAAG... AAGTCCAAG... AAGTCAAG... ATT-CC-AAG... ATT-CC-AAG... ATTTCC-AAG... ATTCCC-AAG... AT--CGGAAG... AT-CCG-AAG... AT-CCA-AAG... AT-CCA-AAG... AATTCC-AAG... AATTCC-AAG... AATTTCCAAG... AATTCCCAAG... AAGTCC-AAG... AAGTCC-AAG... AAGTCC-AAG... AAGTC--AAG...
CLUSTAL CLUSTAL W (1.81) Multiple Sequence Alignments Sequence 1: ArabidopsisAAG52143 798 aa Sequence 2: ArabidopsisAAC26676 845 aa Sequence 3: yeast 664 aa ArabAAG52143 FIVDEADLLLDLGFRRDVEKIIDCLPRQR-------QSLLFSATIPKEVRRVS-QLVLKR 539 ArabAAC26676 FIVDEADLLLDLGFKRDVEKIIDCLPRQR-------QSLLFSATIPKEVRRVS-QLVLKR 586 yeast -VLDEADRLLEIGFRDDLETISGILNEKNSKSADNIKTLLFSATLDDKVQKLANNIMNKK 323 ::**** **::**: *:*.* . * .:. ::******: .:*:::: ::: *: : . * Symbols used?
Binomial distribution • Toss a fair coin once. What is the probability of having a head (H)? • Toss a fair coin twice. What is the probability of having HH, HT or TT (T for tail)?(0.5+0.5)2 • Toss a fair coin 10 times and record the outcome, e.g., HTHHTHHTTT. What is the expected number of HH occurring in the string?E = n p = (10 – 2 +1)0.52 • What is the probability of 0, 1, 2, ..., 9 matches?(p + q)n = (0.25 + 0.75)9
Basic stats in string matching • Given PA, PC, PG, PT in a target (database) sequence, the probability of a query sequence, say, ATTGCC, having a perfect match of the target sequence is:prob = PA (PC)2 PG (PT)2 • Let M be the target sequence length and N be the query sequence length, the “matching operation” can be performed (M – N +1) times, e.g., Query: ATGTarget CGATTGCCCG • The probability distribution of the number of matches follows a binomial distribution with p = prob and n = (M – N +1)
Basic stats in string matching • Computation involving a binomial distribution requires taking the factorial of n. When n is large (e.g., > 1000), this becomes impractical or impossible for today’s computers. • Approximation is therefore necessary. • When np > 50, the binomial distribution can be approximated by the normal distribution with the mean = np and variance = npq • When np < 1 and n is very large (which implies that p is very small given np < 1), binomial distribution can be approximated by the Poisson distribution with mean and variance equal to np (i.e., = 2 = np).
Matching two sequences without gap • Assuming equal nucleotide frequencies, the probability of a nucleotide site in the query sequence matching a site in the target sequence is p = 0.25. • The probability of finding an exact match of L letters is a = pL = 0.25L = 2-2L = 2-S, where S is called the bit score in BLAST. • M: query length; N: target length • The query can start at (M – L +1) positions and the target can start at (N – L +1) positions. These two values are called effective lengths of the two sequences and designated m and n, respectively. They are shorter than M and N, respectively. • The expected number of matches with length L is mn2-S, which is called e-value in ungapped BLAST. • S is calculated differently in the gapped BLAST
Expected number of matches: No gap 123456789012345678901234T: GGTTACACGAGTGCTG ||||||||||||||||Q:CTGAGGTTACACGAGTGCTGCTGA M = 24, L = 16m = M – L + 1 = 24-16+1 = 9E = m2-S = 92-216 =2.09548E-09 1234567890123456789012345T:AACCGGTTACACGAGTGCTGAATGC ||||||||||||||||Q:CTGAGGTTACACGAGTGCTGCTGA M = 25, N = 24, L = 16m = M – L + 1 = 25-16+1 = 10n = N – L + 1 = 24-16+1 = 9 E = mn2-S = 1092-216 =2.09548E-08
Gapped BLAST • Adapted from Crane & Raymer 2003 • Input sequence: AILVPTVIGCTVPT • Algorithm: • Break the query sequence into words:AILV, ILVP, LVPT, VPTV, PTVI, TVIG, VIGC, IGCT, GCTV, CTVP, TVPT • Discard common words (i.e., words made entirely of common amino acids) or sequence of low complexity • Search for matches against database sequences, assess significance and decide whether to discard to continue with extension using dynamic programming:AILVPTVIGCTVPTMVQGWALYDFLKCRAILVPTVIACTCVAMLALYDFLKC
Blast Output (Nuc. Seq.) BLASTN 2.2.4 [Aug-26-2002] ... Query= Seq1 38 Database: MgCDS 480 sequences; 526,317 total letters Score E Sequences producing significant alignments: (bits) Value MG001 1095 bases 34 7e-004 Score = 34.2 bits (17), Expect = 7e-004 Identities = 35/40 (87%), Gaps = 2/40 (5%) Query: 1 atgaataacg--attatttccaacgacaaaacaaaaccac 38 |||||||||| ||||||||||| |||||| |||||||| Sbjct: 1 atgaataacgttattatttccaataacaaaataaaaccac 40 Lambda K H 1.37 0.711 1.31 Matrix: blastn matrix:1 -3 Gap Penalties: Existence: 5, Extension: 2 … effective length of query: 26 effective length of database: 520,557 Matches: 35*1 = 35 Mismatches: 3*(-3) = -9 Gap Open: 1*5 = 5 Gap extension: 2*2 =4 R = 35 - 9 - 5 - 4 = 17 S = [λR – ln(K)]/ln(2) =[1.37*17-ln(0.711)]/ln(2) = 34 E = mn2-S = 26 * 520557 * 2-34 = 7.878E-04 x p(x) 0 0.999265217 1 0.000734513 2 0.000000270 3 0.000000000
E-Value in BLAST • The e-value is the expected number of random matches that is equally good or better than the reported match. It can be a number near zero or much larger than 1. • It is NOT the probability of finding the reported match. • Only when the e-value is extremely small can it be interpreted as the probability of finding 1 match that is as good as the reported one (see next slide).
Problem with BLAST Q: GGC GCG CCC AAG CUG UGC ......T: GGT GCA CCT AAA CUA UGT ...... Alternatives: FASTA AA sequence