Download
1 / 28
280 likes | 461 Views
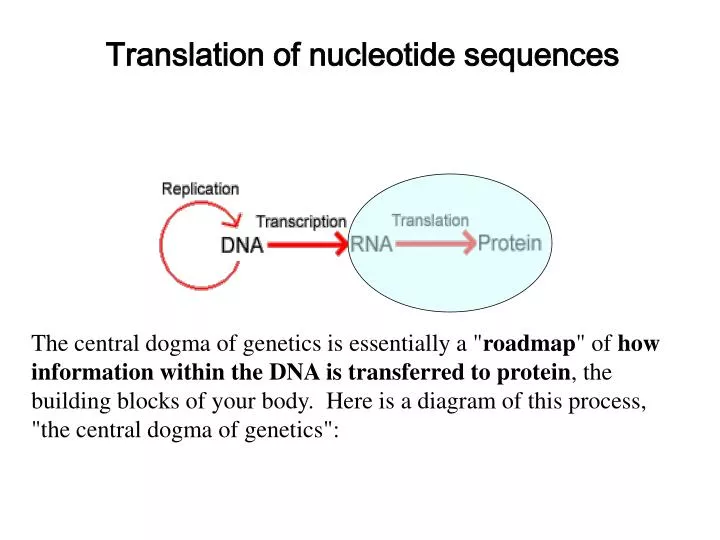
Translation of nucleotide sequences. The central dogma of genetics is essentially a " roadmap " of how information within the DNA is transferred to protein , the building blocks of your body. Here is a diagram of this process, "the central dogma of genetics":.

E N D
Translation of nucleotide sequences The central dogma of genetics is essentially a "roadmap" of how information within theDNA is transferred to protein, the building blocks of your body. Here is a diagram of this process, "the central dogma of genetics":
The DNA-helix is composed of two strands (called watson and crick strands) complementary to each other and genes occur on both strands. The genes are always transcribed in 5’ - 3’ direction. Once a gene has been sequenced it is important to determine the correct open reading frame (ORF). Every region of DNA has six possible reading frames, three in each direction. An open reading frame starts with an atg (Met) in most species and ends with a stop codon (taa, tag or tga).
In the W2H file window, select the file ab001874. Select the program MAP, and click start. Clostridium paraputrificum gene for chitinase B, complete cds (coding sequence).
Q. Based on the results of translation, what are the differences between the codes of mold mitochondria and the standard code? Name one of these differences. • Different genetic codes are described here: • The Standard Code • The Vertebrate Mitochondrial Code • The Yeast Mitochondrial Code • The Mold, Protozoan, and Coelenterate Mitochondrial Code and the Mycoplasma/Spiroplasma Code • The Invertebrate Mitochondrial Code • The Ciliate, Dasycladacean and Hexamita Nuclear Code • The Echinoderm and Flatworm Mitochondrial Code • The Euplotid Nuclear Code • The Bacterial and Plant Plastid Code • The Alternative Yeast Nuclear Code • The Ascidian Mitochondrial Code • The Alternative Flatworm Mitochondrial Code • Blepharisma Nuclear Code • Chlorophycean Mitochondrial Code • Trematode Mitochondrial Code • Scenedesmus Obliquus Mitochondrial Code • Thraustochytrium Mitochondrial Code
Select the sequence mito.seq in the file list. The sequence is from a mitochondrial gene for a mold cytochrome c oxidase. The standard code UUU Phe F UCU Ser S UAU Tyr Y UGU Cys C UUC Phe F UCC Ser S UAC Tyr Y UGC Cys C UUA Leu L UCA Ser S UAA Stop* UGA Stop* UUG Leu L UCG Ser S UAG Stop* UGG Trp W CUU Leu L CCU Pro P CAU His H CGU Arg R CUC Leu L CCC Pro P CAC His H CGC Arg R CUA Leu L CCA Pro P CAA Gln Q CGA Arg R CUG Leu L CCG Pro P CAG Gln Q CGG Arg R AUU Ile I ACU Thr T AAU Asn N AGU Ser S AUC Ile I ACC Thr T AAC Asn N AGC Ser S AUA Ile I ACA Thr T AAA Lys K AGA Arg R AUG Met M ACG Thr T AAG Lys K AGG Arg R GUU Val V GCU Ala A GAU Asp D GGU Gly G GUC Val V GCC Ala A GAC Asp D GGC Gly G GUA Val V GCA Ala A GAA Glu E GGA Gly G GUG Val V GCG Ala A GAG Glu E GGG Gly G
The Standard Code The Mold, Protozoan, and Coelenterate Mitochondrial Code Differences from the Standard Code: The Mold...Standard TGA Trp W Ter * Alternative Initiation Codons: Trypanosoma: TTA, TTG, CTGLeishmania: ATT, ATA Tertrahymena: ATT, ATA, ATG Paramecium: ATT, ATA, ATG, ATC, GTG, GTA(?) (Pritchard et al., 1990)
Identification of open reading frames Q. Name some of the protein products (FT: CDSs) of the ectrmd operon. Select the sequence ectrmd( E. coli trmD operon and nearby regions ) in the file list,which is an operon. FT CDS 416. .1522 >> /note="unidentified reading frame FT CDS 1771. .2019 >> /note="ribosomal protein S16 (rps P gene) (aa 1-82)” FT CDS 2617. .3384 >> /note="tRNA (m1G) methyltransferae (trmD gene) (aa 1-255)” FT CDS 3426. .3773 >> /note="ribosomal protein L19 (rplS gene) (aa 1-115)" FT CDS complement(3849. .4232) >> /note="unidentified reading frame (16K polypeptide) (aa1-127)"
Comparing two sequences - local and global alignment You will now compare the two protein sequences 1.pep and 2.pep using the "Gap" program (Gap Protein). Q. What is the quality value when comparing 1.pep and 2.pep? What quality values are obtained with 10 randomisations? May we conclude that the alignment corresponds to a pair of evolutionary related proteins? Q. Are the results from "Gap" and "Bestfit" different? What alignment is more significant?
GAP BESTFIT
Aligning two sequences - Gap extension penalty. Alignment of genomic sequence with mRNA Select the following two sequences: V00594 (Human mRNA for metallothionein) and J00271 (corresponding genomic sequence). Q. Based on the alignment, how many exons are there in this gene? Compare your result to what's in the annotation section for J00271. Default settings Open gap= 50 Extend gap= 3
New settings Open gap= 10 Extend gap= 0
Comparing two sequences based on exact matching of short words Compare the two sequences 1.pep and 2.pep with COMPARE.
Identification of repeats in a human chromosome analyze the sequence chr21.seq with COMPARE. Q. What is the relationship between the number of diagonals in the plot and the number of repeats? Approximately how many nucleotides are there in the repeated sequence? 5 4 3 2 1 A
Searching databases for sequencehomology BLAST search in the W2H environment. In this exercise we will identify homologs of the 54 kDa subunit of the mammalian signal recognition particle (SRP54).Q. Name one mammalian ortholog to the mouse srp54 according to the blast output.
BLAST and the role of the substitution matrix in the detection of remote similarity. Go to BLAST at the NCBI. 1. VFQQDNDPKHTSLHVRSWFQRRHVHLLDQPSQSPDLNPIEH 2. IFLHDNAPSHTARAVRDTLETLNWEVLPHAAYSPDLAPSDY First use the default matrix BLOSUM62, then the PAM250 matrix. Q. The remote similarity between these two proteins is detected only with one of the matrices. Which one? Score = 31.2 bits (69), Expect = 4.9Identities = 14/41 (34%), Positives = 22/41 (53%) Query: 1 VFQQDNDPKHTSLHVRSWFQRRHVHLLDQPSQSPDLNPIEH 41 +F DN P HT+ VR + + +L +SPDL P ++ Sbjct: 1 IFLHDNAPSHTARAVRDTLETLNWEVLPHAAYSPDLAPSDY 41
BLAST and sequence filtering. Use the protein with accession P19934(E. coli TolA protein). Q. From the result of the search with filtering: Give an example of a region filtered in the query sequence (examine the alignments)?Give an example of a sequence with a high score that is found in the output without filtering but is not found in the output with filtering?
Multiple sequence alignment PILEUP Apply pileup on a set of related proteins from two different families, sr54 and ftsy. Q. What protein family (sr54 or ftsy) has a C-terminal portion* which is lacking in the other family ? Q. What protein family has a N-terminal portion which is lacking in the other family ? Q. Can you identify a highly conserved sequence motif in the alignment, i.e a subsequence shared by all members of the the srp54 and ftsy protein family ? * Protein sequences have a polarity, the N- (NH2 or amino-) terminus is at the left and the C-(COOH or carboxy-) terminus at the right in a linear sequence. This is sometimes denoted like: N-GHYTRWYYQIIQ-C where "GHYTRWYYQIIQ" is the actual amino acid sequence. A concept like "the N-terminal half" of this (very short) protein would then be the sequence "GHYTRW". Proteins are typically made up of two or more functional domains. The SRP54 and Ftsy proteins are examples of this. The multiple sequence alignment allows us to identify the differences between the proteins with respect to domain organisation.
N-terminal C-terminal
A search in pfam (www.sanger.ac.uk/Software/Pfam/) confirm our findings. Sr54_arcfu Ftsy_aquae
Pattern searches Pattern recognition Cx{2,4}Cx12Hx{3,5}H The search pattern defined means: A cysteine residue followed by any two to four amino acids, a cysteine residue, any 12 amino acids, histidine, any three to five amino acids and finally histidine. ). Q. Give an example of a human protein in Swissprot described as a zinc finger protein and that is identified with findpatterns! ! FINDPATTERNS on SwissProt:* allowing 0 mismatches ! Using patterns from: zincfinger September 22, 2003 13:31 .. ABF1_HUMAN ck: 1650 len: 3,703 ! Q15911 homo sapiens (human). alpha-fetoprotein enhancer bindingprotein (a zinc_zinger Cx{2,4}Cx12Hx{3,5}H Cx{2}Cx{12}Hx{4}H 284: KPILM CFLCKLSFGYVRSFVTHAVHDH RMTLS
2.Search Swissprot human proteins for the sequence "GDSGGP. Examine the result carefully.Are you able to identify serine proteases? Are there proteins that are not proteases? ! FINDPATTERNS on SwissProt:* allowing 0 mismatches ! Using patterns from: serineproteases September 22, 2003 14:09 .. 2ACC_HUMAN ck: 2269 len: 414 ! Q9y5p8 homo sapiens (human). serine/threonine protein phosphatase 2a, 48 k serine_proteases GDSGGP 331:SLLRD GDSGGP ELSDW ACH1_LONAC ck: 3441 len: 213 ! P23604 lonomia achelous (giant silkworm moth) (saturnid moth). achelase i serine_proteases GDSGGP 186: RDQCQ GDSGGP LYHNR ACH2_LONAC ck: 5072 len: 214 ! P23605 lonomia achelous (giant silkworm moth) (saturnid moth). achelase ii serine_proteases GDSGGP 187:RDQCQ GDSGGP LYHNG Q62284 ck: 7458 len: 135 ! Q62284 mus musculus (mouse). gamma-7s nerve growth factor (y-ngf) (fragmen serine_proteases GDSGGP 85: KDTCK GDSGGP LICDG
3. Using FINDPATTERNS, identify human Swissprot proteins that have stretches of at least 40 consecutive serine residues ( hint, use S{40,} as search pattern. Q. Give one example of a protein that has least 40 consecutive serines (strange protein!). ! FINDPATTERNS on SwissProt:*_HUMAN allowing 0 mismatches ! Using patterns from: serine40 September 22, 2003 15:31 .. AF9_HUMAN ck: 6118 len: 568 ! P42568 homo sapiens (human). af-9 protein. 2/2003 pat_ S{40,} S{40} 149: RSIHT SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS SSTSF S{40} 150: SIHTS SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS STSFS S{40} 151: IHTSS SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS TSFSK Databases searched: SWISS-PROT, Release 60.0, Released on 24Mar2003, Formatted on 24Mar2003 Total finds: 3 Total length: 4,795,187 Total sequences: 9,172 CPU time: 42.26
Motifs Search for protein motifs in the Swissprot sequence fa7_human. Q. What motifs are found with MOTIFS? MOTIFS from: /d/u/paul/work/fa7_human.pep Mismatches: 0 September 22, 2003 15:34 .. fa7_human.pep Check: 4382 Length: 466 ! ID FA7_HUMAN STANDARD; PRT; 466 AA. ______________________________________________________________________________ Asx_Hydroxyl Cx(D,N)x4(F,Y)xCxC Cx(D)x{4}(Y)xCxC 121: QNGGS CKDQLQSYICFC LPAFE **************************************************** Aspartic acid and asparagine hydroxylation site *************************************************** Post-translational hydroxylation of aspartic acid or asparagine [1] to form erythro-beta-hydroxyaspartic acid or erythro-beta-hydroxyasparagine has been identified in a number of proteins with domains homologous to epidermal growth factor (EGF). Examples of such proteins are the blood coagulation protein factors VII, IX and X, proteins C, S, and Z, the LDL receptor, thrombomodulin, etc. Based on sequence comparisons of the EGF-homology region that contains hydroxylated Asp or Asn, a consensus sequence has been identified that seems to be required by the hydroxylase(s). -Consensus pattern: C-x-[DN]-x(4)-[FY]-x-C-x-C [D or N is the hydroxylation site] -Note: this consensus pattern is located in the N-terminal of EGF-like domains, while our EGF-like cysteine pattern signature (see the relevant entry <PDOC00021>) is located in the C-terminal. -Last update: January 1989 / First entry. [ 1] Stenflo J., Ohlin A.-K., Owens W.G., Schneider W.J. J. Biol. Chem. 263:21-24(1988).