Download

1 / 30
350 likes | 892 Views
Jodie Ouahed , MD Victor Fox, MD Boston Children’s Hospital Reviewed by Diana Riera , MD of the Professional Education Committee. Polyposis. Common presentations of polyposis syndromes:. Rectal Bleeding (often painless) Abdominal Pain Altered bowel habit Prolapse of polyp/rectum

E N D
Jodie Ouahed, MD Victor Fox, MD Boston Children’s Hospital Reviewed by Diana Riera, MD of the Professional Education Committee Polyposis
Common presentations of polyposis syndromes: • Rectal Bleeding (often painless) • Abdominal Pain • Altered bowel habit • Prolapse of polyp/rectum • Intussusception • Asymptomatic
Classification and diagnostic considerations: Classification: • 1) Hamartomas • 2) Adenomas • 3) Inflammatory • 4) Mixed polyposis syndromes • 5) Hyperplastic Most commonly seen: • -Solitary: Solitary Juvenile Polyp (hamartoma) • -Familial: Familial Adenomatous Polyposis Diagnosis of pediatric Polyposis Syndromes: • Requires clinical presentation with thorough physical exam including • Macrocephaly • Penile freckling • Unusual lentigines (pigmented macules on lips, mouth, face, extremities, café au lait macules ) • Histologic findings • Once detected careful Family History (FHx) must be obtained including history of cancer, and age of onset
Familial Adenomatous Polyposis: • Hundreds-thousands of adenomas in the colon, rectum as well as stomach and small intestines • Most of the gastric polyps are non-neoplasticfundic gland type polyps, but can develop foci of dysplasia or adenoma • The duodenum is frequently involved while more distal portions of the small bowel are rarely involved • Begin in childhood/adolescence • Increase in number with age • Clinical dx of classic FAP: if >100 colorectal adenomatous polyps identified • Attenuated FAP (8%): Milder form with fewer adenomas and later presentation; less likely to present in childhood
Epidemiology & Presentation FAP: • Epidemiology: 1/100 000 births • 20-30% are spontaneous mutation • Pre-symptomatic presentation: for genetic testing (affected family member) • Symptomatic: with multiple colonic adenomas • Typically identified in teenage years, at the time of screening (not usually following symptoms) • Most have easily detectable colorectal adenomas by late childhood-early adolescence • Age of presentation is debatable (depends on when scope is performed) • Some found as young as 4-5 y/o
Signs and Symptoms of FAP: • GI manifestations: • Often asymptomatic, until advanced neoplasia occurs • Increased frequency of bowel movements • Looser stools • Mucous discharge • Rectal bleeding • Abdominal or back pain. • Extra-intestinal manifestations: • Hepatoblastoma • Thyroid and adrenal carcinoma • Teeth: impacted, supernumerary, uninterrupted teeth • Connective tissue: desmoid tumors, subcutaneous cysts, fibromas, excessive adhesions • Eyes: Benign asymptomatic retinal lesions called congenital hypertrophy of retinal pigment epithelium (CHRPE)
Pathophysiology: Genetics • Autosomal Dominant • Mutated APC (Adenomatous Polyposis Coli) geneon chromosome 5q21, usually results in truncated protein • APC is a tumor suppressor gene; thus inactivation of one APC allele predisposes to adenoma formation • Phenotypic variability with: • Location of mutation: Most severe between codons 1250-1464, especially codon 1309. • Environmental factors • Other modifier genes • Autosomal recessive form exists (mutated MUTYH gene) • This does not present in childhood
Screening and Diagnosis of FAP: • Genetic testing for APC in at risk relatives is recommended just before 10-12 y/o • Controversy on whether to test for APC earlier, given rare risk of hepatoblastoma (~1%) , which usually presents within first 5 years of life • Diagnosis is confirmed by finding polyps on flex sig/colonoscopy, histologically confirmed as adenomas • Assure detailed counseling prior to screening • Anticipate psychological, family, insurance, and employment implications • Have clear plan in place for post-test management
Screening and Diagnosis of FAP: • Mutations may only be detected in 70-90% of cases! • A) If mutation not found: Can’t offer predictive testing • Require endoscopic surveillance • Protocols vary, but annual sigmoidoscopy on all first degree relatives until adenomas are found • Starting at 20 y/o colonoscopy with dye spray q5 years • B) If mutation identified: directed testing is used to predict risk of FAP in relatives: • Absence of this gene mutation – excludes FAP; relatives are at average population risk for adenomas/cancer • Presence of gene mutation confirms diagnosis of FAP - requires endoscopic assessment
Management of FAP: • At risk children need a screening flexible sigmoidoscopy q1-2yrs by 10-12 yrs old • Earlier if early aggressive disease in family
Management of FAP continued: • Colectomy indicated if large # of adenomas, or adenomas with high degree of dysplasia • Colectomy is only means to eliminate inevitable risk of colorectal cancer • Severe dysplasia and carcinoma reported in < 12 y/o • Surgical options: • IPAA preferable if large # of rectal adenomas (>15-20), >1000 colonic adenomas, high risk genotypes, or if severe dysplasia
FAP prognosis: • Almost all will develop colorectal cancer by 5th decade, if not detected and treated at an early age • Average age of colorectal cancer is 39 y/o • In pediatric population, polyps in stomach and small intestine don’t usually require intervention • Adults at higher risk of cancer of duodenum, Ampulla of Vater. • Risk of Hepatoblastomas in children < 5y/o • Higher risk of other cancers: thyroid (in adolescence) brain, pancreas • Higher risk of death from extra-colonic manifestations (desmoid tumor, duodenal cancer)
FAP Variants • Gardner Syndrome • Familial colorectal polyposis • Extracolonic tumors include osteomas of the skull, thyroid cancer, epidermoid cysts, fibromas, sebaceous cysts, and desmoid tumors • Turcot Syndrome • Brain tumor-polyposis syndrome • Association of familial polyposis of the colon with brain tumors including medulloblastoma and malignant glioma Celiac Disease
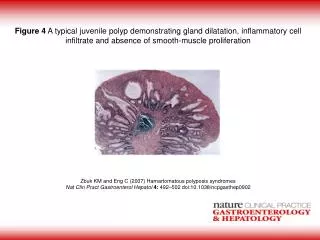
Juvenile Polyp: Typical presentation • Solitary hamartoma at diagnosis • Mean age of presentation: 4 y/o • Painless rectal bleeding • Perianal polyp protrusion • Up to 40% children have multiple polyps; 60% proximal to rectosigmoid
Juvenile Polyp: Management: • Requires full colonoscopy, because if multiple polyps, 60% are proximal to rectosigmoid • Polyps should be removed, even if incidental finding • If solitary polyp after full colonoscopy, and no relevant family history, polypectomy is sufficient
Juvenile Polyp: Prognosis: • A) If solitary- no increased risk of colorectal cancer, though polyp can recur in patients who present with a solitary polyp • B) If recurrence of symptoms- it may represent a first presentation of hamartamotous polyposis syndrome; so always re-investigate! • C) If multiple juvenile polyps or a +FHx of colonic polyps or colorectal cancer, must consider juvenile polyposis syndrome
Juvenile Polyposis Syndrome (JPS): Epidemiology and Presentation • 1/100 000 individuals • Multiple juvenile-type hamartomatous polyps • Increased risk of GI malignancies • Consider JPS if: • >5 juvenile polyps in colon • Juvenile polyps in other parts of GI tract • Any # of juvenile polyps in a patient with a positive family history
JPS: Pathophysology • Autosomal Dominant • Fully penetrant; Variable expression • 60% familial; the rest are sporadic • Germline mutations in SMAD4 (20%), BMPR1A (20%), and possibly ENG1. Some include PTEN mutations in this description too. • SMAD4 mutation is a tumor suppressor gene, and predisposes to gastric polyps and hereditary hemorrhagic telangiectasia
JPS management: • Once a gene mutation is identified, test at risk family members • If gene mutation known: - Genetic testing, after appropriate counseling. - If SMAD4 mutation, should also screen for HHT (Hereditary hemorrhagic telangiectasia) • If gene mutation unknown: -Screen first degree children with colonoscopy by 12 y/o -Colonoscopic surveillance q2years for children • JPS patients surveillance EGD and full colonoscopy q2-3 years starting at 15 y/o; earlier if polyps are clinically apparent • Colectomy is warranted if cancer, high-grade dysplasia, or high polyp burden can’t be controlled endoscopically
Peutz-Jerghers Syndrome (PJS): • 1/50 000-1/200 000 live births • Muco-cutaneous pigmentation and hamartomatous polyps throughout GI tract • Polyps are mostly in SI (especially jejunum), less in stomach and colon • Presumptive dx if positive family history and typical freckling • Patients may have polyps in kidneys, bladder, lungs, and nares
PJS presentation: • Polyps can cause bleeding, anemia, SI intussusception with intestinal obstruction at young age • Pigmentation occurs in infancy • Sites of pigmentation: on lips and inside mouth, nostrils, perineum, hands and feet- including fingers and toes • Pigmentation may fade after puberty, but persists in buccal mucosa
Clinical diagnosis and pathophysiology of PJS: • Any child/adolescent with any one of: • 2 or more histologically confirmed PJ polyps • Any # of PJ polyps and positive family history in close relative(s) • Characteristic mucocutaneous pigmentation and positive family history in close relative(s) • Any # of PJ polyps in an individual with characteristic mucocutaneous pigmentation • Autosomal Dominant • Mutated STK11 gene (tumor suppressor) is found in 90% patients • Marked phenotypic variability between and within families.
Management of PJS: • If mutation identified, test at risk relatives after genetic counseling • Midgut complications require polypectomy, either by deep enteroscopy (eg. double or single balloon technique), or laparotomy/laparoscopy and intra-operative enteroscopy (IOE) • Management of asymptomatic polyps: • Polyps < 1.0 cm, no symptoms → counsel about risk of intususception • Polyps >1.5cm → consider removal with deep enteroscopy or IOE and screen q2-3 years with endoscopy, colonoscopy and wireless capsule endoscopy or MRE • Polypectomy should be only performed by experienced experts!
Screening for polyps and malignancies in patients with PJS • Baseline EGD and colonoscopy by 8 y/o • If polyps present: q3yrs till 50 y/o • If no polyps: repeat at 18 y/o, then q3 yrs till 50 y/o • Small bowel should be assessed by wireless capsule endoscopy or MRI enterography q3yrs from 8 y/o • Annual clinical exam, Hgb, liver function tests • Annual exam of genital tract (testicular U/S q2yrs from birth till 12 y/o; cervical smear q3 yrs from 25 y/o) • Monthly self breast exam from 18 y/o; annual breast MRI from 25-50, then yearly mammography
Prognosis of PJS: • 60% affected will require laparotomy in childhood • High re-operation rate after first laparotomy for SI obstruction presenting at young age • Relative risk of cancer is 15.2 compared to rest of population, majority are in GIT and occur in adulthood • Tumors in GI tract: colon, pancreas, stomach • Extraintestinal tumors: lung, testes, ovary, breast,uterus, cervix
PTEN Hamartoma Tumor Syndrome • Group of genetic syndromes each associated with mutated PTEN tumor suppressor gene. • Rare 1/200 000; 50% de novo mutations • Phenotypic variants include: • Cowden syndrome: Autosomal dominant • 90% have small hamartomatous colonic polyps distal to hepatic flexure. • Rarely presents in children. associated with macrocephaly, acralkeratosis, • Bannayan-Riley-Ruvalcaba Syndrome: Autosomal Dominant • Presents before adolescence • Associated with Macrocephaly, developmental delay, lipomatosis, hemangiomatosis, increased risk of cancer (renal, thyroid, breast) • Proteus Syndrome: • Hemi-hypertrophy, but few GI issues
References: • Barnard J. Screening and Surveillance Recommendations for Pediatric Gastrointestinal Polyposis Syndromes. Journal of Pediatric Gastroenterology and Nutrition. 2009. 48 (Suppl 2) S75-S78 • Fox V. Juvenile polyps: recurrence in patients with multiple and solitary polyps. Clinical gastroenterology and hepatology 2010 8(9):795-799 • Beggs et al. PeutzeJeghers syndrome: a systematic review and recommendations for management. Gut. 2010 Jul;59(7):975-86. • Large genomic deletions of SMAD4, BMPR1A and PTEN in juvenile polyposis. Van hattem et al. Gut. 2008 May;57(5):623-7. • Wyllie, Hyams and Kay. Pediatric Gastrointestinal and Liver Disease. Fourth ed, Ch. 43: p462-471.