Download

1 / 47
470 likes | 516 Views
This review delves into the clinical presentation, genetic features, and gross anatomy of Huntington's Disease. Explore its impact on motor, cognitive function, and mood, as well as how mutated huntingtin gene alters normal cellular functions, leading to neurodegeneration. Gain insights into disease models, inhibitors, and potential therapeutics.

E N D
Exam 2 Review: Neurodegenerative Diseases Biology of Mental Disorders
Huntington’s Disease Lecture 1: Clinical Presentation • Trinucleotide repeat disease • Autosomal dominant • Genotype=Phenotype • Affects men and women across all races/ethnic groups, but most common in people of European descent • Motor impairments: Chorea(“dance-like” movements) uncontrolled, jerky movement of arms, legs, head, upper trunk • Cognitive impairments: impairments in reasoning skills, memory, concentration, judgment, organization ability • Mood changes: depression, anxiety, anger/irritability
Huntington’s Disease Lecture 1: general genetic features • Mutation in huntingtin gene (HTT) on chromosome 4 • Huntingtin has polyglutamine “polyQ” triplet “CAG” codon repeat sequence • Repeat creates “sticky”, large intracellular aggregates • Expansions due to DNA polymerase “slipping” events • Anticipation • Juvenile HD: >60 repeats • Adult HD: >36 repeats
Huntington’s Disease Lecture 1: Gross Anatomy • Progressive neurodegeneration • Huge ventricles, severe striatal/basal ganglia atrophy, general gray matter thinning • Loss of GABA-ergic, medium spiny neurons (MSN’s) in striatum • May target striatum due to glutamate overaccumulation, leading to excitotoxicity • increased glu release increased Ca2+increased neuron death
Direct and Indirect Pathways: Huntington’s HUNTINGTON’S NORMAL FUNCTIONING Cortex Cortex Putamen D2 D1 Thalamus Thalamus Putamen D2 D1 Direct pathway Direct pathway SNc SNc Indirect pathway Indirect pathway GPe GPe Gpi/SNr Gpi/SNr Red = Inhibitory Green = Excitatory STN STN
Huntington’s Disease Lecture 2: Gain of Function • Gain of function features of Htt gene: expanded polyQ enables protein to do something it wouldn’t normally do • Higher levels of mHTT protein correlate with disease phenotypes • Expression of polyQ HTT fragments is toxic • Gene knockout in mice results in different anatomical and early life abnormalities that aren’t seen in people with the expanded allele Mice with different expression levels of same 120Q expansion: Higher protein expression less time on rotarod
Huntington’s Disease Lecture 2: Caspase 6 • Expression of cleaved caspase6fragments can result in disease • Cleavage fragments decreased latency to fall • Caspase-6 activity increased with mHTT expression in mice • Mutated caspase-6 cleavage site (C6R) is less harmful (looks like WT) Caspase 6 fragment with 86 Q repeat (C63) mice Caspase 6 fragment with 23 Q repeat (A2) mice Control mice
Huntington’s Disease Lecture 2: Loss of Normal Function • Loss of function features of Htt gene: polyQ prevents protein from doing what it normally does • Htt KO is lethal • Heterozygous mice are hyperactive & have cognitive deficits • Homozygosity for expansion is indistinguishable from heterozygosity in humans and not lethal in mice (expansion affects adult functions not developmental functions)
Huntington’s Disease Lecture 2: How Expanded Htt alters normal functions • Apoptosis: • Expanded Htt prevents cells from undergoing apoptosis when exposed to oxidant • Excitotoxicity: • Expanded Htt Inability to restore resting membrane potential • Expanded Htt Reduced ATP/ADP ratio • Expanded Htt Increased NMDA currents • Expanded Htt cells more susceptible to glutamate excitotoxicity • Altered nuclear function: • Nuclear export sequence aggregates in nucleus (rather than cytoplasm) • Reduction in BDNF • Neuronal transport: • Normal Htt important for shuttling mRNA and proteins & transcriptional regulation (NRSF/REST) • Mitochondrial health: • Expanded Htt Impaired resting mitochondrial membrane potential • Inhibit cytochrome c release with CSA (mitochondria transition core blocker) prevents excitotoxicity
Huntington’s Disease Lecture 2: histone deacetylase (HDAC) inhibitors • Rationale: • Huntingtin protein interacts with histone acetyltransferase (HAT) proteins, like CREB-binding protein (CBP) • CBP is found in nuclear inclusions with mutant htt • Mutant htt • Leads to less histone acetylation, recruitment of HAT complexes to DNA, lower gene expression • Inhibiting deacetylasesmay improve function
Huntington’s Disease Lecture 2: Disease Models • Drosophila eye model • Examine differences in histone acetylation • Large libraries of small molecule screens in C. elegans • Mouse models WT Poly-Q suppressor=hsp70 Mechanistic Therapeutic
Alzheimer’s Disease Lecture 1: Clinical Presentation • Impaired: Complex attention, executive function, learning and memory, language, motor perception and social cognition • Age-dependent (exponential increase after 6th decade of life) • African American and Hispanic individuals at higher risk AAMI MCI Cognitive Performance AD Age
Alzheimer’s Disease Lecture 1: Pathology Tau tangles Amyloid plaque • Pathology most likely begins before diagnosis • Extracellular amyloid plaques & tau tangles • Hippocampal/cortical degeneration (apoptosis, excitotoxicity, mitochondrial dysfunction, calcium dynamics, inflammation, etc…) • AD targets large pyramidal neurons (ex: CA1 and CA3 in hippocampus) • Cerebellum largely spared Asymptomatic Symptomatic Unresponsive substrate Responsive substrate?
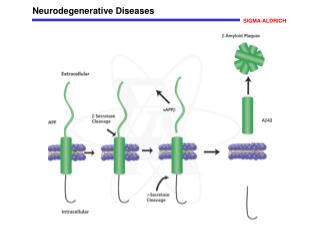
Alzheimer’s Disease Lecture 1: Pathology (continued) • Abeta accumulation: • Reduced clearance • Reduced degradation (Ubiquitin proteasome pathway) • Inflammation • Mutations in APP, PS1, PS2 • APOE4allele (APOE2 is protective) • α-,β-, and γ-secretase: • α=good, β=bad, γ=neutral • Most issues with Abeta42, issues with Abeta40; go on to form oligomers and plaques
Alzheimer’s Disease Lecture 2: Cholinergic Hypothesis • Basal forebrain cholinergic (CBF) cell death correlates with disease severity, pathology, synapse loss • CBF neurons depend on NGF for survival/maintenance • Hypothesis: inability to obtain or use NGF underlies demise of CBF neurons • AD may prevent retrograde transport of NGF in cholinergic neurons (decreased NGF receptors & ACh+ neurons with age) • Reduced NGF signaling may lead to Abeta accumulation
Alzheimer’s Disease Lecture 2: Amyloid hypothesis • Aβ: • Primary component of senile plaques, disrupt neuronal function neuronal death • May shift caspase-3 activity with age from “plasticity path”(protective factor) to a “cell death path” (pathogenic factor) • May spread in a “prion-like” manner • Gain of function
Alzheimer’s Disease Lecture 2: Disease Models • Amyloid precursor protein (APP) important for: • Axonal transport • Cell adhesion • Cell growth • Synaptic plasticity • Mutations in APP, PS1, and PS2are hallmarks offamilial AD • Have Abeta plaques, loss of synapses, spatial memory impairments, gliosis • Combining APP + PS1 earlier onset • APP KO mice indistinguishable from WT (implies plaques, etc… = gain of function) • Triplicate mouse model (3xTg) has beta secretase, APP, and familial mutation in tau gene • LTP deficits (weaker LTP with age)
Alzheimer’s Disease Lecture 2: BACE and HDAC inhibitors • BACE (beta-site APP-Cleaving Enzyme) inhibitor • beta secretase inhibitor • Reduces Abeta production • BACE KO mice have decreased pathology • BACE KO rescues memory defect in mice • HDAC (histone deacetylase) inhibitor • Preserves learning and memory performance in mice • Restores “lost” memory in mice
Alzheimer’s Disease Lecture 3 Journal: Extrasynaptic NMDAR in AD • Synaptic NMDAR: • Activated by low-frequency stimulation by glutamate released from vesicles • Activate CREB, stimulate BDNF, neuroprotective • More a-secretase, less abeta • Extrasynaptic NMDAR: • Activated by spillover glutamate at dendrite shaft, cell body • Decrease CREB, BDNF, dysregulate mitochondria, glutamate toxicity • Less a-secretase, increased abeta, & increased BACE • Importance of where glutamate enters
Alzheimer’s Disease Lecture 3 Journal: Extrasynaptic NMDAR in AD • Memantine/Nitromemantine: targets extrasynaptic NMDAR’s • Decrease abeta levels • Decrease phospho-tau levels • Blocks ROS & mitochondrial Ca2+ overload • Prevents CREB shut off • Questions: • Does A-beta stimulate glutamate release & where? Is it synaptic or extrasynaptic? • What happens in post-synaptic neurons after glutamate release? • Does A-beta affect synaptic plasticity? • What happens in spines after a-beta activation of eNMDAR?
Alzheimer’s Disease Lecture 3 Journal: Extrasynaptic NMDAR in AD • Does A-beta stimulate glutamate release and where? Is it synaptic or extrasynaptic? • It engages α-7 nicotinic acetylcholine receptors to release astrocytic glutamate extrasynaptic NMDA receptors on neurons • What happens in post-synaptic neurons after glutamate release? • Increases in calcium and nitric oxide • Chronic A-beta exposure in vivo shows increased glu baseline current • Does A-beta affect synaptic plasticity? • In hippocampus, eNMDAR activity is followed by reduction in evoked and miniature excitatory postsynaptic currents (mEPSC’s) • What happens in spines after a-beta activation of eNMDAR • Greater presence of phosphorylated tau • In vivo synapse loss • Memantine and nitromemantine rescue functions of above features
Alzheimer’s Disease Lecture 3 Journal: Extrasynaptic NMDAR in AD
Alzheimer’s Disease Journal Club: REST overexpression • REST: • Chromatin modifying silencing factor • Represses neurogenesis in embryonic development • Expressed in NSC’s; diminished when neurons mature • Overexpression blocks migration and neuronal differentiation • Goals: • Create conditional REST overexpression knock-in mouse • Determine role of REST • Determine relationship between REST and DRD2 gene • Measure locomotion
Alzheimer’s Disease Journal Club: REST overexpression • Made knock-in mouse • Cre-loxp model • Gene that REST regulates • See genome shift (from genes modifying postsynaptic membrane potential to gene modifying regulation of immune response) • REST seems to target DRD2 • Strong binding of DRD2 in striatal neurons • Relationship between REST and DRD2 • REST represses DRD2 • Locomotor findings • Increased immobility, decreased speed, and decreased total distance traveled by REST overexpression + DRD2 mice • Locomotion findings not due to neuronal or glial cell death/morphology changes, or apoptosis—confirms that locomotor findings are due to relationship between REST and DRD2
Parkinson’s Disease Lecture 1: Clinical Presentation • Chronic, progressive neurodegenerative disease that primarily affects movement • Age-dependent (60 yo; young-onset = <40 yo) • More frequent in men • Highly variable severity • Not immediately fatal; die from complications • Cardinal Motor Features: • (Resting) Tremor • Rigidity • Akinesia/bradykinesia • Postural Instability • Non-motor symptoms: • Anosmia • Depression/anxiety • Cognitive problems (focused attention, planning, language/memory) • Digestive issues • Fatigue • Sleep disorders
Parkinson’s Disease Lecture 1: Pathology • Progressive loss of dopaminergic neurons in the substantia nigra (SN) & nigrostriatal tract • α-synuclein (and ubiquitin +) deposits called Lewy Bodies aggregate as a result of dysfunction of ubiquitin proteasome system and lysosomal autophagy Lewy Body
Direct and Indirect Pathways: Parkinson’s PARKINSON’S NORMAL FUNCTIONING Cortex Cortex Putamen D2 D1 Putamen D2 D1 Thalamus Thalamus X Direct pathway Direct pathway SNc SNc Indirect pathway Indirect pathway GPe GPe Gpi/SNr Gpi/SNr Red = Inhibitory Green = Excitatory STN STN
Parkinson’s Disease Lecture 1: Genetic Causes • Mutation on chromosome 4 at SCNA gene(codes for α-synuclein) • Results from point mutations, triplication • α-synuclein present at presynaptic vesicles and promotes vesicle docking and fusion • Can form lipid-based aggregates • Excess α-synuclein can be released extracellularly—too much α-synuclein can be endocytosed by neighboring cells, leading to propagation • LRRK2, also Parkin, DJ-1, PINK-1, and ATP13A2 • Mutations in GBA(glucosylceramidasebeta) gene (important for lysosome function and autophagy) promotes α-synuclein propagation
Parkinson’s Disease Lecture 1: Environmental Causes • Herbicides (paraquat) • Pesticides (rotenone, permethrin, organochlorines) • Rural environments • Consuming well water • Proximity to industrial plants/quarries • Metal exposure (high-dose manganese & chronic lead exposure)
Parkinson’s Disease Lecture 2: MPTP Models • Neurotoxin MPTP mimics pathophysiology of PD byselectively targeting DA neurons in SN; inhibiting complex 1 of mitochondria reduction in TH+ neurons • Chronic MPTP administration: • Lewy bodies (α-synuclein & ubiquitin deposits) • Gliosis • Behavioral deficits • Apomorphine (APO): dopamine receptor agonist that rescues loss of DA neurons due to MPTP
Parkinson’s Disease Lecture 2: Genetic Models • Point Mutations causing accelerated aggregation of α-synuclein • A53T (more potent) • A30P • Overexpression • α-synuclein triplication (SCNA X 3) • Gain of function • Point mutation/overexpression models (rodentand fly) show motor deficiencies
Parkinson’s Disease Lecture 2: α-synuclein neurobiology • α-synuclein KO protective against MPTP • Effects of α-synuclein aggregates: • Disrupts proteosome activity; increases susceptibility to protein degradation • Disrupts mitochondrial respiration (complex I dysfunction increased ROS cytochrome c release apoptosis) • Binds to and depletes chaperones • Exacerbates dopamine-induced cell death • Altered vesicular recycling
Parkinson’s Disease Lecture 2: PD genes • PINK1 andParkinwork together to induce mitophagy, mutations here cause issues • LRRK2: important for synaptic vesicle formation; KO’s have fewer and smaller vesicle size with altered vesicular recycling
Parkinson’s Disease Lecture 2: Current Treatments and neuroprotective features • Most common 1st line of treatment: L-DOPA • Combined with carbidopa to prevent metabolism outside of brain and increase efficiency/availability of L-DOPA crossing BBB • Stem cell transplants • Deep brain stimulation • Thin wire electrode in STN or Gpi that delivers electrical pulses • Blocks over activation of Gpi/SNr • Neuroprotective features: • Nicotine • BDNF treatment • Exercise
Parkinson’s Disease Journal Club: Gut microbiota • 80% of PD patients have GI symptoms • Microbiome: bacteria that live in gut/intestines • Diversity is key • Question: Do peripheral symptoms in PD connect to findings in brain? • Vagus nerve • How does gut microbiome influence brain function in PD mouse models? • Goals: • Do gut microbiomes effect locomotor behavior? • What is the effect of gut microbiome on α-synuclein pathology? • Are there signs of CNS inflammation caused by microbiome? • Are effects of gut microbiome something determined in early life or is it an active, lifelong communication?
Parkinson’s Disease Journal Club: Gut microbiota • Do gut microbiomes effect locomotor behavior? • alpha synuclein overexpression (ASO) without gut bacteria (GF): • locomotion and GI movements like wild-type mice • Short-chain fatty acids (SCFA’s) seem to “mediate” PD gut-brain signaling • What is the effect of gut microbiome on α-synuclein pathology? • GF-ASO mice have limited/no α-synuclein deposits; same α-synuclein production as SPF-ASO mice, but different handling • Signs of inflammation caused by microbiome? • GF-ASO mice microglia look like wt (more branches, more branching, more “space-filling”) • Are effects of gut microbiome something determined in early life or is it an active, lifelong communication? • Active, lifelong communication • Human fecal transplants: mouse resembled phenotype of human donor