Download

1 / 60
660 likes | 893 Views
Enzyme Kinetics. 9/1/2004. General Properties of Enzymes. Increased reaction rates sometimes 10 6 to 10 12 increase Enzymes do not change D G just the reaction rates. Milder reaction conditions Great reaction specificity Can be regulated. Substrate specificity.

E N D
Enzyme Kinetics 9/1/2004
General Properties of Enzymes • Increased reaction rates sometimes 106 to 1012 increase • Enzymes do not change DG just the reaction rates. • Milder reaction conditions • Great reaction specificity • Can be regulated
Substrate specificity • The non-covalent bonds and forces are maximized to bind substrates with considerable specificity • Van der Waals forces • electrostatic bonds (ionic interactions) • Hydrogen bonding • Hydrophobic interaction • Stereospecificity • Geometrically specific • A + B P + Q • Substrates Products enz
Enzymatic catalysis and mechanisms • A. Acid - Base catalysis • B. Covalent catalysis • C. Metal ion aided catalysis • D. Electrostatic interactions • E. Orientation and Proximity effects • F. Transition state binding • General Acid Base • Rate increase by partial proton abstraction by a Bronsted base or • Rate increase by partial proton donation by a Bronsted Acid
Enzyme Kinetics • Rates of Enzyme Reactions • How fast do reactions take place • Reaction rates • Thermodynamics says I know the difference between state 1 and state 2 and DG = (Gf - Gi) • But • Changes in reaction rates in response to differing conditions is related to path followed by the reaction • and • is indicative of the reaction mechanism!!
Chemical kinetics and Elementary Reactions A simple reaction like A B may proceed through several elementary reactions like A I1 I2 B Where I1 and I2 are intermediates. The characterization of elementary reactions comprising an overall reaction process constitutes its mechanistic description. Rate Equations Consider aA + bB + • • • + zZ. The rate of a reaction is proportional to the frequency with which the reacting molecules simultaneously bump into each other
The order of a reaction = the sum of exponents Generally, the order means how many molecules have to bump into each other at one time for a reaction to occur. A first order reaction one molecule changes to another A B A second order reaction two molecules react A + B P + Q or 2A P
3rd order rates A + B + C P + Q + R rarely occur and higher orders are unknown. Let us look at a first order rate A B n = velocity of the reaction in Molar per min. or moles per min per volume k = the rate constant of the reaction
Instantaneous rate: the rate of reaction at any specified time point that is the definition of the derivative. We can predict the shape of the curve if we know the order of the reaction. A second order reaction: 2A P Or for A + B P + Q
Percent change in A (ratio ) versus time in first and second order reactions
It is difficult to determine if the reaction is either first or second order by directly plotting changes in concentration.
However, the natural log of the concentration is directly proportional to the time. - for a first order reaction- The rate constant for the first order reaction has units of s-1 or min-1 since velocity = molar/sec and v = k[A] : k = v/[A] Gather your data and plot ln[A] vs time.
The half-life of a first order reaction Plugging in to rate equation The half-life of a first order reaction is the time for half of the reactant which is initially present to decompose or react. 32P, a common radioactive isotope, emits an energetic b particle and has a half-life of 14 days. 14C has a half life of 5715 years.
A second order reaction such like 2A P When the reciprocal of the concentration is plotted verses time a second order reaction is characteristic of a straight line. The half-life of a second order reaction is and shows a dependents on the initial concentration
The Transition State A bimolecular reaction A + B C A B + C at some point in the reaction coordinate an intermediate ternary complex will exist A B C This forms in the process of bond formation and bond breakage and is called a transition state. Ha + Hb Hc Ha Hb + Hc This is a molecule of H2 gas reforming by a collision
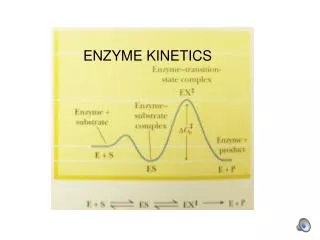
An energy contour of the hydrogen reaction as the three molecules approach the transition state at location c. This is called a saddle point and has a higher energy than the starting or ending point.
Energy diagrams for the transition state using the hydrogen molecule Transition state diagram for a spontaneous reaction. X‡ is the symbol for the species in the transition state
For the reaction Where [X] is the concentration of the transition state species ‡ ‡ k' = rate constant for the decom-position of the activated complex ‡ ‡ ‡ DG‡ is the Gibbs free energy of the activated complex.
‡ The greater the DG‡,the more unstable the transition state and the slower the reaction proceeds. This hump is the activation barrier or kinetic barrier for a reaction. The activated complex is held together by a weak bond that would fly apart during the first vibration of the bond and can be expressed by k' = kn where n is the vibrational frequency of the bond that breaks the activated complex and k is the probability that it goes towards the formation of products.
Now we have to define n. E = hn and n = E/h where h is Planks constant relating frequency to Energy. Also through a statistical treatment of a classical oscillator E= KbT where Kb is Boltzmann constant. By putting the two together ‡ And The rate of reaction decreases as its free energy of activation, DG‡ increases or the reaction speeds up when thermal energy is added
Multi-step reactions have rate determining steps Consider If one reaction step is much slower than all the rest this step acts as a “bottleneck” and is said to be the rate-limiting step
Preferential transition state binding Binding to the transition state with greater affinity to either the product or reactants. RACK MECHANISM Strain promotes faster rates The strained reaction more closely resembles the transition state and interactions that preferentially bind to the transition state will have faster rates kN for uncatalyzed reaction and kE for catalyzed reaction
‡ ‡ ‡ ‡ ‡ ‡ ‡ ‡ ‡ ‡ ‡ ‡ ‡ ‡
106 rate enhancement requires a 106 higher affinity which is 34.2 kJ/mol The enzyme binding of a transition state (ES‡ ) by two hydrogen bonds that cannot form in the Michaelis Complex (ES) should result in a rate enhancement of 106 based on this effect alone
Catalysts act to lower the activation barrier of the reaction being catalyzed by the enzyme. Where DDG‡cat = DG‡uncat- DG‡cat The rate of a reaction is increased by DDG‡cat = 5.71 kJ/mol is a ten fold increase in rate. This is half of a hydrogen bond!! DDG‡cat = 34.25 kJ/mol produces a million fold increase in rate!! Rate enhancement is a sensitive function of DDG‡cat
Kinetics of Enzymes Enzymes follow zero order kinetics when substrate concentrations are high. Zero order means there is no increase in the rate of the reaction when more substrate is added. Given the following breakdown of sucrose to glucose and fructose Sucrose + H20 Glucose + Fructose
E = Enzyme S = Substrate P = Product ES = Enzyme-Substrate complex k1 rate constant for the forward reaction k-1 = rate constant for the breakdown of the ES to substrate k2 = rate constant for the formation of the products
When the substrate concentration becomes large enough to force the equilibrium to form completely all ES the second step in the reaction becomes rate limiting because no more ES can be made and the enzyme-substrate complex is at its maximum value. [ES] is the difference between the rates of ES formation minus the rates of its disappearance. 1
Assumption of equilibrium k-1>>k2 the formation of product is so much slower than the formation of the ES complex. That we can assume: Ks is the dissociation constant for the ES complex.
Assumption of steady state Transient phase where in the course of a reaction the concentration of ES does not change 2
3 Combining 1 + 2 + 3 rearranging Divide by k1 and solve for [ES] Where
vo is the initial velocity when the reaction is just starting out. And is the maximum velocity The Michaelis - Menten equation
The Km is the substrate concentration where vo equals one-half Vmax
The KM widely varies among different enzymes The KM can be expressed as: As Ks decreases, the affinity for the substrate increases. The KM can be a measure for substrate affinity if k2<k-1
There are a wide range of KM, Vmax , and efficiency seen in enzymes But how do we analyze kinetic data?
Lineweaver-Burk plot: slope = KM/Vmax, 1/vo intercept is equal to 1/Vmax the extrapolated x intercept is equal to -1/KM For small errors in at low [S] leads to large errors in 1/vo kcat is how many reactions an enzyme can catalyze per second The turnover number
For Michaelis -Menton kinetics k2= kcat When [S] << KM very little ES is formed and [E] = [E]T and Kcat/KM is a measure of catalytic efficiency
What is catalytic perfection? When k2>>k-1 or the ratio is maximum Or when every substrate that hits the enzyme causes a reaction to take place. This is catalytic perfection. Then Diffusion-controlled limit- diffusion rate of a substrate is in the range of 108 to 109 M-1s-1. An enzyme lowers the transition state so there is no activation energy and the catalyzed rate is as fast as molecules collide.
Kinetic data cannot unambiguously establish a reaction mechanism. Although a phenomenological description can be obtained the nature of the reaction intermediates remain indeterminate and other independent measurements are needed.
Reaction MechanismsA: Sequential Reactions • All substrates must combine with enzyme before reaction can occur
Ping-Pong Reactions • Group transfer reactions • One or more products released before all substrates added
Inhibition kinetics • There are three types of inhibition kinetics competitive, • mixed and uncompetitive. • Competitive- Where the inhibitor competes with the substrate.