Download

1 / 1
10 likes | 129 Views
Transition metal-free catalytic hydrogenation of ketones Katherine Jolley and Martin Wills Department of Chemistry, The University of Warwick, Coventry, CV4 7AL, UK. Introduction.

E N D
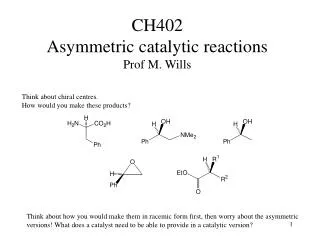
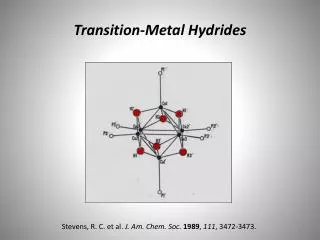
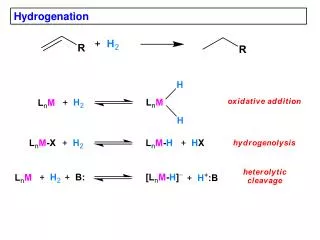
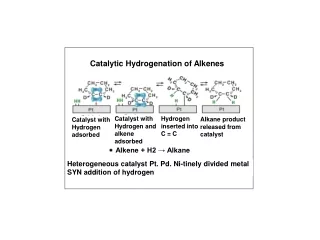
Transition metal-free catalytic hydrogenation of ketones Katherine Jolleyand Martin Wills Department of Chemistry, The University of Warwick, Coventry, CV4 7AL, UK Introduction Hydrogenation is an important chemical process used in the production of a vast range of chemical products. A large number of homogeneous and heterogeneous transition metal hydrogenation catalysts have been developed, however literature suggests that little work has been carried out in the development of catalysts based on cheaper and more readily available non-transition metals1 . The only precedent for such a system dates back to 1964 when Walling and Bollyky reported the base-catalysed hydrogenation of ketones, focussing on the hydrogenation of benzophenone to benzhydrol using KOtBu base as the catalyst2 as shown in Scheme 1 (right). Scheme 1. Hydrogenation of benzophenone to benzhydrol using KOtBu catalyst. Further investigations3,4 into the reaction proposed a mechanism involving a 6-membered cyclic transition state as shown in Figure 1 (left) which is analogous to the ruthenium system developed by Noyori.5 We propose that the use of tetradentateligands in conjunction with an alkali metal base may help induce order upon the system and force the approach of the ketone substrate from a specific orientation (Figure 1) which would allow the reaction to proceed under milder conditions. Figure 1. (Left) Proposed 6-membered transition state for KOtBu catalysed hydrogenation of benzophenone. (Centre) Mechanism for Noyori’s system. (Right) Proposed mechanism involving multidentate ligands. Results and Discussion 2 Work began with repetition of the reaction developed by Walling and Bollyky2 and the identification of appropriate catalyst and ligand loadings as shown in Table 1 (right) Little improvement in conversion was seen upon increasing the catalyst loading from 20 to 40 mol% (Entries 2 and 3) and thus 20 mol% was selected. Triethylene glycol was initially used as the initial ligand and it was found that matching the ligand and catalyst loadings as in Entry 6 gave the highest conversion. It should also be noted that adding the ligand gave a 2% increase in conversion cf. Entry 2 and 6. Work then proceeded to investigate the effectiveness of other tetradentateligands with differing elements at the 4 co-ordination sites. Ligand 2 (bis-(2-hydroxyethyl)ethylenediamine) was the most effective. The results are shown below in Figure 2. 3 1 2 4 Figure 2. % conversions to benzhydrol achieved with various tetradentate ligands. 0.4 M benzophenone, 20 mol % KOtBu, 20 mol% ligand, 60 bar H2, 60°C, tBuOH. a Conversion determined by gas chromatography. Table 1. Identification of reaction conditions Work continued by investigating the use of solvents with different boiling points in order to help reduce levels of solvent evaporation over the long reaction times. Table 2 (left) summarises the results. The use of 2-methyl-2-butanol gave similar conversions to tBuOH, but significantly less evaporation was observed. Triethylene glycol is thought to fully saturate the potassium and prevent the reaction from taking place. The addition of ligand in Entries 2 and 4 significantly increases conversion to benzhydrol. With improved reaction conditions more tetradentateligands with ligand 2’s backbone were synthesised and tested. The results are shown below in Figure 3. Ligands5 and 6 were not effective and it is thought that the phenol OH groups do not co-ordinate to the potassium as well as the free OH groups in ligands1, 7, 8 and 9. 5 6 8 7 9 a Conversion determined by gas chromatography. Reactions carried out at 60°C, 60 bar H2, with 0.4 M benzophenone. Figure 3. % conversion to benzhydrol achieved with various tetradentate bis-hydroxy diamine ligands. 0.4 M benzophenone, 20 mol% KOtBu, 20 mol% ligand, 60 bar H2, 60°C, 2-methyl-2-butanol. Table 2. Investigating the use of different solvents. Conclusions and proposed reaction mechanism The results shown above in Figure 3 for ligands7-9 allowed a reaction mechanism to be proposed as shown in Figure 4 (right). Initially it was thought that the tert-butoxide base interacts with the nitrogen proton via hydrogen bonding. Once held close to the system the base can then interact with molecular hydrogen and bring it close to the ketone which is co-ordinated to the potassium. This then allows for efficient hydride transfer from H2 to the ketone. With ligands7 and 9, the nitrogen is part of a secondary amine and therefore has a proton which can interact with the base. Ligand 8 however has a tertiary amine, with a methyl instead of a proton. The methyl hydrogens will not interact with the base and thus the hydrogen molecule cannot be brought close to the ketone. The progress of the reaction will be hindered explaining the lack of conversion to product for ligand 8. Figure 4. Proposed mechanism for hydrogenation of benzophenone using ligands 7 and 8. However…. Analysis of the reaction solution after the reaction using ligand 9 was found to show both mono and fully oxidised ligand. It was thought that the mechanism may involve hydrogen transfer from the ligand to the ketone rather than reaction of the ketone with molecular hydrogen. Indeed reactions using ligand 9 carried out under atmospheric nitrogen rather than pressurised H 2 showed similarconversion. It is thought that the mechanism may be similar to that seen with Meerwein-Ponndorf-Verley systems6 and Oppenauer Oxidation systems7 as shown in Figure 5 (left). The location of the phenyl group adjacent to the hydroxy group in ligands7 and 9 is thought to promote such a hydrogen transfer process by allowing formation of a more conjugated and hence more stable system upon oxidation of the alcohol. Work continues to further investigate the reaction mechanism. Figure 5. (left) Mechanism for Oppenauer Oxidation7, (right) proposed mechanism using KOtBu and ligand 7. References 1. P. Chase, G. Welch, T. Jurca and D. Stephan, Angew. Chem. Int. Ed., 2007, 46, 8050-8053. 2. C. Walling and L Bollyky, J. Am. Chem. Soc., 1964, 86, 3750-3752. 3. A. Berkessel, T. Schubert and T. Muller, J. Am. Chem. Soc., 2002, 124, 8693-8698. 4. B Chan and L Radom, J. Am. Chem. Soc., 2005, 127, 2443-2454. 5. R. Noyori, M. Yamakawa, S. Hashiguchi, J. Org. Chem., 2001, 66, 7391-7944. 6. C. R. Graves, E. J. Campbell, S. T. Nguyen, Tetrahedron: Asymmetry., 2005, 16, 3460-3468. 7. R. Mello, J. Martinez-Ferrer, G. Asensio, M. E. Gonzalez-Nunez, J. Org. Chem., 2007, 72, 9376-9378. Acknowledgements I would like to thank Martin Wills and the Wills group for their help and support during this project and the EPSRC for funding of the project.