Download

1 / 1
10 likes | 171 Views
A. Regulatory region. Terminator region. Regulatory region. orf 1. orf 4. orf 2. orf 3. orf 5. Coding region. Co-transcribed genes. Upstream region. 4. Prediction Reliability

E N D
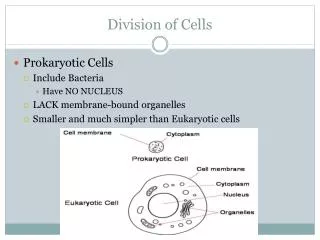
A Regulatory region Terminator region Regulatory region orf 1 orf 4 orf 2 orf 3 orf 5 Coding region Co-transcribed genes Upstream region 4. Prediction Reliability One of the main advantages provided by PREDetector is the opportunity for the user to estimate the reliability of the predictions. The large natural occurrence of transcription factors binding sites are located within intergenic regions and not within coding sequences. PREDetector provides these statistics and therefore the user can estimate the scores at which he will find strongly or weakly reliable sites. 2. Regulon Prediction The search for potential binding sites of the regulatory protein starts with the selection of one of the saved weight matrices and the definition of the cut-off score. The lowest score among the input sequences used to build a matrix is fixed by default as the recommended cut-off score for this matrix. Users can modify the cut-off score. PREDetector is able to scan either complete or selected regions of bacterial genomes available in the GenBank database. Users can determine the bounds of the so-called “regulatory regions” (estimation of maximal distances upstream and downstream the translational start wherein functional regulatory motifs could be found), as well as bounds of co-directionally transcribed genes. PREDetector : Prokaryotic Regulatory Element Detector Samuel Hiard1, Sébastien Rigali2, Séverine Colson2, Raphaël Marée1 and Louis Wehenkel1 1Bioinformatics and Modeling, GIGA & Department of Electrical Engineering and Computer Science – University of Liège, Sart-Tilman B28, Liège, Belgium 2Centre for Protein Engineering – University of Liège, Sart-Tilman B6 Liège, Belgium Abstract Background: In the post-genomic area, in silico predictions of regulatory networks are considered as a powerful approach to decipher and understand biological pathways within prokaryotic cells. The emergence of position weight matrices based programs has facilitated the access to this approach. However, a tool that automatically estimates the reliability of the predictions and would allow users to extend predictions in genomic regions generally regarded with no regulatory functions was still highly demanded. Result: Here, we introduce PREDetector, a tool developed for predicting regulons of DNA-binding proteins in prokaryotic genomes that (i) automatically predicts, scores and positions potential binding sites and their respective target genes, (ii) includes the downstream co-regulated genes, (iii) extends the predictions to coding sequences and terminator regions, (iv) saves private matrices and allows predictions in other genomes, and (v) provides an easy way to estimate the reliability of the predictions. Conclusion: We present, with PREDetector, an accurate prokaryotic regulon prediction tool that maximally answers biologists’ requests. PREDetector can be downloaded freely at http://www.montefiore.ulg.ac.be/~hiard/predetectorfr.html The weight matrix based approach Transcription factor binding sites are usually slightly variable in their sequences. Positional weight matrix summarizes information about binding sites sequence alignment. It also allows to predict the occurrence of new sites and estimate their binding efficiency for a transcription factor. The generation of a position weight matrix starts with the alignment of the experimentally validated DNA motifs of a specific transcription factor. Multiple alignment A C G T C A C G G T C C G C T The multiple alignment is then converted into an alignment matrix that represents how many times nucleotide i was observed in position j of the alignment. The alignment matrix is then converted into a weight matrix via the formula: where : - ni,jis the observed frequency of nucleotide i in position j - N is the number of sequences in the set - pi is the expected frequency of nucleotide i in the genome. For instance 0,25 for each nucleotide in a 50% rich GC genome. Weight matrix Scores in red are those for the best nucleotide at each position. The consensus sequence is ACG(C/G)T. The score of a L-length sequence is computed by summing the weights of each nucleotide. Why PREDetector ? Our motivation to generate PREDetector came from our intense utilisation of previously described similar programmes, such as Target Explorer (A. Sosinsky et al., 2003), Predictregulon (S. Yellaboina et al, 2004), or Virtual footprint (R. Munch et al., 2006), that were not appropriate to predict some of our in vivo experimentally validated DNA binding sites. The priority and challenge of PREDetector was to offer a programme which, all at once, would provide an easy way to estimate the reliability of the predictions, and beyond the identification of strongly reliable cis-acting elements, would guarantee users the possibility to access information among the predicted sites with scores generally regarded with no regulatory function because categorized beyond statistical reliability thresholds. 3. Results Once the options have been set, PREDetector scans the selected genome sequences and classifies the predicted target DNA motifs according to their localisation in the genome. This includes coding sequences or intergenic sequences, which can be classified as (1) regulatory regions (where regulatory elements are predicted to be found), (2) upstream regions (any region upstream of a translational start codon), and (3) terminator regions (in PREDetector a terminator region terminology is only used to indicate regions between two translational stop codons). Predictions results are distributed among these four genome localization categories 1. Weight matrix creation The first part of PREDetector consists in the generation of a weight matrix according to a set of experimentally validated binding sites. The weight matrix can be saved into user’s library and further used to scan different bacterial genomes. Conclusion PREDetector is an accurate prokaryotic regulon prediction tool that maximally answers biologists’ requests. Suggestions for improvements are welcome (contact S.Hiard@ulg.ac.be, L.Wehenkel@ulg.ac.be).