Download

1 / 16
160 likes | 351 Views
Amino Acid Scoring Matrices. Jason Davis. Overview. Protein synthesis/evolution Computational sequence alignment Smith-Waterman Algorithm BLAST Amino Acid Scoring Matrices PAM – Point Accepted Mutations BLOSUM – BLOck SUbstitution Matrix mPAM Metric Conversions. Proteins.

E N D
Amino Acid Scoring Matrices Jason Davis
Overview • Protein synthesis/evolution • Computational sequence alignment • Smith-Waterman Algorithm • BLAST • Amino Acid Scoring Matrices • PAM – Point Accepted Mutations • BLOSUM – BLOck SUbstitution Matrix • mPAM • Metric Conversions
Proteins • 3-dimensional stuctures • Composed of amino acids chained together • Can be represented as a 2-dimensional sequence • 20 different amino acids exist • Usually 100-1500 amino acids long • Have many different shapes and functions • Function depends on both 3d shape and aa sequence
Protein Synthesis • DNA: strand composed of 4 different base pairs • A, T, C, G • 20 amino acids: 3 base pairs needed to encode each amino acid • Degenerate coding Protein Signalling Transcription/Translation
Protein Evolution • Protein ‘families’ • Set of homologous proteins • Same function, different composition • Similar structure • Identifying families • Pairwise sequence alignment • Multiple sequence alignment • NP-hard • Other approaches • Structural, experimental
Pairwise Sequence Alignment • Input • 2 sequences p, q of lengths m,n • 20x20 Amino Acid Substitution Matrix • Insertion (gap) cost • Global Alignment • Find optimal set of insertions such that the resulting alignment (length < m+n) is optimal w.r.t. amino acid substitution matrix • Difficult, less useful • Local Alignment • Find significant ‘hotspot’ in the alignment
Sequence Alignment Algorithms • Dynamic Programming Approaches • Global and Local variations • Provably Optimal • O(nm) space and time • ‘banded’ heuristics can reduce the state space • FSA extensions allow varying penalties for gap openings and gap extensions • Heuristics Approaches • Blast, Fasta • Sublinear time – look for statistical significance in small local alignments between sequences
Substitution Matrices - PAM • Dayhoff, Schwartz, Orcutt (1978) • Step 1: extrapolate mutation probabilites from 1 step in evolutionary time • Pick a set of protein families (71) • Restrict proteins in each family to sequences with similarity above a certain threshold (>85%) • Build a phylogenetic tree for each family • Extrapolate frequencies Aab that amino acids a, b evolved from same amino acid • Aab and Aba assumed to be the same • Convert frequencies to probabilities • p(a|b) = Bab = Aab/∑cAac
Substitution Matrices – PAM (2) • Step 2 – Infer greater evolutionary times • Dayhoff defined a PAM1 matrix to have 1% expected substitutions • For each row, scale off-diagonalsand adjust diagonals to keep the matrix row stochastic • To infer larger evolutionary times, we can view formed matrix C as a 20-state Markov Chain • Cn is the result of performing n-steps in the Markov Process
Substitution Matrices – PAM (3) • Create odds ratio of • 1) the event that 2 amino acids i,j, evolved from the same ancestor, x • fi = observed frequency of amino acid i • p(i,j have same ancestor) = ∑xfx Pr{x→i} Pr{x→j} = ∑xfx (CN)ix (CN)jx= ∑x (CN)ix fx (CN)jx= ∑x (CN)ix fj (CN)xj= fj (C2N)ij • 2) the event that the 2 amino acids align at random • p(independent alignment of i,j) = fi * fj • Final log odds ratio: • Dij = average[log((CN)ij / fi), log(CN)ji / fj)) • The log allows for an additive model • Final numbers are rounded to nearest integer
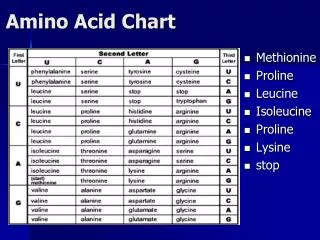
PAM250 • Different values on the diagonal correspond do mutability potential
BLOSUM • Henikoff & Henikoff, 1992 • Uses aligned, ungapped blocks within protein families that have similarity greater than some level L% • qa = ∑bAab / ∑c,d Acd • pab = Aab / ∑c,d Acd • S(a,b) = log(pab / qaqb) • Final entries are rounded • Blosum62 (L=62), Blosum50 (L=50) • More direct approach, usually yields better results
Log-Odds Similarity Matrix Properties • Negative numbers needed for Smith-Waterman local alignment algorithm • Nice probabilistic interpretation • Amino acid substitutions assumed independent • Attempts to metricize these matrices • Taylor, Jones 93: used various algebraic manipulations to arrive at a metric matrix with minimal disortion • Dij = a – Sij • Larger values of a yielded better metrics at the cost of high dimensionality • Constant Shift Embedding • Linial, et. al. constructed a near metric over aligned segments of length 50 • D(u,v) = S(u,u) + S(v,v) – 2*S(u,v) • 10-7error rate
mPAM • Metric substitution model • Measures the expected time per 250 mutations among 100 amino acids • Same rate as PAM250 • Exponential distribution assumed: f(t) = 1 – e-λt • Given pairwise substitution rates p(a,b) • Solve for λ: f(1) = 1-e- λ = p(a,b) • Expected time t of an event occuring in an exponential distribution is 1/ λ • mPAM(a,b) = round(1/ λ) • Two values needed to be adjusted to form a metric • Rounding error?
mPAM (2) • Seller’s Theorem: • If a pairwise alignment is found using a metric, resulting alignment scores are also metrics • Optimized for BLAST-like lookup • Smaller alignments • Difficult to compare with other similarity matrices • Dynamic programming algorithms rely on negative values in the similarity matrix • Probabilistic interpretation: larger positive alignments are statistically significant
mPAM Disadvantages • d(x,x) = 0 • This does not capture the relative mutability among different amino acids • PAM/BLOSUM capture this with different positive values along the diagonal • Do amino acids substitute according to an exponential distribution? • Amino Acid Substitution may be inherently non-metric • Comparison to BLOSUM?