Download

1 / 34
530 likes | 1.39k Views
Scoring matrices. Identity PAM BLOSUM. Scoring Matrices Types. Identity matrix – exact matches receive one score and non-exat matches a different score (say 1 and 0, or 6 and –1 for local alignment.).

E N D
Scoring matrices Identity PAM BLOSUM
Scoring Matrices Types • Identity matrix – exact matches receive one score and non-exat matches a different score (say 1 and 0, or 6 and –1 for local alignment.). • Mutation data matrix – a scoring matrix compiled based on observation of protein point mutation (PAM, BLOSUM). • Physical properties matrix – amino acids with with similar properties (e.G. hydrophobicity ) receive high score. • Genetic code matrix – amino acids are scored based on similarities in the coding triple (codons).
Substitution Matrix • Amino acids substitute easily for another due to similar physicochemical properties • Isoleucine for Valine (both small, hydrophobic) • Serine for Threonine (both polar) • Such changes – “conservative” • Thus, need a way to increase sensitivity of the alignment algorithm • Solution – substitution matrix • Therefore, we need a range of values that depend on the nature of sequences being compared • Identical amino acids > Conservative substitutions > Nonconservative substitutions
Choice of scoring matrix is dictated by the alignment goals • Two proteins are homologous if (and only if) they are evolutionarily related (have a common ancestor) • Homologous proteins are likely to have related functions (and have the same fold) • Scoring matrices must in some way model our understanding of protein evolution. • Based on the result of the search we have to be able to decide if the discovered sequence similarity could happen by chance or is a signature of likely homology.
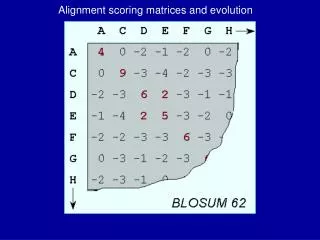
BLOSUM • Block – a short contiguous interval of multiple aligned sequences. • BLOCKS – data base of 3 000 blocks of highly conserved sequences representing hundreds of protein groups. • Http://www.Blocks.fhcrc.Org/. • BLOCKS substitutions frequency log odds score. • Within each block cluster sequences within certain similarity threshold (80% similarity yields BLOSUM80) and have such cluster be represented by one sequence or average the contribution. • BLOSUM62 – most similar to PAM250 (believed to be better).
Deriving a frequency tables from a data base of blocks Computing a logarithm of odds matrix 1.2 7.5 6.3 1.9 5.5 3.1 6.5 2.0 8.1 4.3 3.7 5.8 2.9 7.7 3.2 BLOSUM METHOD Data Base of blocks Data base
Methods Deriving a frequency table from a data base of blocks. Frequency table consisting of all possible amino acid pairs in a column • 9A + 1S there are 8+7+…+1=36 AA pairs • 9 AS or SA pairs • no SS pairs For a block : width of w and a depth of S, it contribute WS(S-1)/2 [1.10.(10-1)]/2=45
METHODS • The result of this counting is a frequency table listing the number of time each of the 20+19+…+1=210 different amino acid pairs occurs among the blocks. • The table is used to calculate a matrix representing odds ratio between these observed frequency and those calculated by chance.
METHODS • Observed probability qij : fAA= 36, fAS= 9 qAA= 36/45 = 0.8 qAS= 9/45 = 0.2
Methods • Expected probability eij : pA= [36 + (9/2)]/45 = 0.9 pS = [00 + (9/2) /45 = 0.1 • for i=j eij = pi.pj ; • eAA = pA.pA = 0.9 x 0.9 = 0.81 • for ij eij = pi.pj + pi.pj ;= 2 pi.pj • eAS = pA.pS + pA.pS = 2 pA.pS = 2 (0.9 x 0.1) = 0.18
Methods • The odds ratio • An odds ratio matrix is calculated where each entry is qij/eij • The logarithm of odds ratio (Lod) in bit unit • Sij = log2qij/eij • A Lod is then calculated as score • If the observed frequency is : • as the expected, then Sij = 0 • if less than expected Sij < 0 • if more than expected Sij > 0
METHODS • Clustering segment within blocks • Sequences are clustered within blocks, and each cluster is weighted. This is done by specifying a clustering percentage in which sequence segments that are identical for at least that percentage of amino acids are grouped together. • The lod matrix derived from a database of blocks in which sequences that are identical at 80% of aligned residues are clustered is referred to as BLOSUM 80, and so forth.
The Dayhoff Matrix (PAM) • Developed by Margaret Dayhoff, 1978. • Counted likelihood of all possible substitutions in closely related proteins. • Derived mutability matrix Mi,j: • Probability that Ai mutates to Aj in one evolutionary unit, PAM. • Multiplying M by itself extrapolate to higher evolutionary orders (Mk).
PAM units • Log-odds approach: Scores proportional to the log of the ratio of target frequencies to background frequencies • PAM – Point Accepted Mutation /Percent Accepted Mutation • Two sequences S and T are defined to be one PAM unit divergedif a series of accepted point mutation (and no insertion/deletion) can convert S to T with an average of one mutation per 100 res. • Point accepted mutation – mutation of one residue accepted by evolution.
PAM units • Problem 1: given two sequences you cannot tell their PAM distance in the strict sense of the above definition since one residue could mutate more than once • BUT: If you take sequences that are closely related then problem above is unlikely to occur. • Problem 2 : A change could happen by deletion/insertion
PAM Matrices - Summary • There is a sequence of PAM matrices • PAMn attempts to provide proper scoring for sequences that diverged n PAM units. • PAMn matrix is obtained from PAM1 assuming Markov model of protein evolution where transition probabilities in 1 PAM step are given by PAM1. • PAMn = PAM1 n • PAM1 is constructed based on highly similar sequences (believed to be apart at most few PAM units) so that Problems1 & 2 are unlikely to occur.)
Computation representation • Define: • fp(a) = probabilities of occurrence for each amino acid a. • f(a,b) = the number of times the mutation a↔b ( f(a,b) = f(b,a) ) • f(a) = b∑f(a,b) ( b≠a ) • m(a) = mutability of amino acid a = f(a) / fp(a)
Computation representation ,cnd • M(a,b) = the probability of amino acid a changing to amino acid b • M(a,b) = Pr(a↔b) = Pr(a↔b | a changed)Pr(a changed) = f(a,b)* m(a) / f(a) (the conditional probability above is estimated as the ratio between the a↔b mutations and the total number of mutations involving a ) • M(a,a) = 1- m(a) unchange probability (the diagonal elements)
Relatedness odds Matrix • M(a,b) gives the probability that amino acid a will change to b in a related sequence in a interval • f(b) is the chance of a random occurrence of amino acid b • Score(a,b) = 10log[M(a,b)/f(b)] (symmetric matrix)
PAM • Let us assume to AA (or nucleotides) i and j, with frequency fiand fj. • P(random alignment of i and j)=fi fj.
Long Distance Evolution • There is a different mutation probability matrix for each evolutionary interval. These can be derived from the one for 1 PAM by matrix multiplication. • e.g. in 2 PAM units of evolution a→c→b (c can be anything including a or b) • In general Mⁿ is the transition probability matrix for a period of n units of evolution
Estimation of Evolutionary Distance • Different mutation probability matrix for each evolutionary interval measured in PAMs. • Calculate the percentage of amino acids that will be observed to change on the average in the interval P = 100(1 – ∑f(i)M(i,i)) • A PAM250 matrix usually represents two sequences which have about 20% identity
Nucleotide PAM scoring matrices Assuming equal probability for each mutation PAM1 would be: A T G C A .99 .0033 .0033 .0033 T .0033 .99 .0033 .0033 G .0033 .0033 .99 .0033C .0033 .0033 .0033 .99 Some models would score higher transitions (purine into purine pirimidine into pirimidine) that transversions: A T G C A .99 .0002 .0006 .0002 T .0002 .99 .0002 .0006 G .0006 .0002 .99 .0002C .0002 .0006 .0002 .99
Discrimination of real local alignment from “by chance” alignment Method: Compute mutual information: Sx Syp(x,y) log(p(x,y)/ p(x)p(y)) Recall that score s(x,y) = log(p(x,y)/ p(x)p(y)) Thus we simply compute: Sx=1..20 Sy=1,..20 p(x,y) s(x,y) Examples (in bits): PAM160 = .7; PAM250 = .36 Higher mutual information better discrimination between true and by chance alignment.
Problems with PAM • Defining PAM 1 in terms of amino acid mutation rather than number of nucleotide changes. • Some mutation may be rare and underrepresented in PAM1 (which is based on closely related proteins only). • The mutation rate depends on the position of an amino-acid in the structure. • Require construction phylogenic tree which in turn need scoring matrices for proper construction. (remains a problem for many other methods)
Some more problems with PAM Matrices • Derived from global alignments of closely related sequences. • Matrices for greater evolutionary distances are extrapolated from those for lesser ones. • The number with the matrix (PAM40, PAM100) refers to the evolutionary distance; greater numbers are greater distances. • Does not take into account different evolutionary rates between conserved and non-conserved regions.
BLOSUM matrices • BLOcks SUbstitution Matrix • Amino acid substitution matrices from protein blocks S. HENIKOFF and J. HENIKOFF Proc. Natl. Acad. Sci.USA Vol.89, pp. 10915-10919, November 1992 Biochmistry
Comparison to PAM • The BLOSUN series derived from alignments in blocks is fundamentally different from the Dayhoff PAM series, which is derived from the estimation of mutation rates. • Nevertheless, the BLOSUM series based on percent clustering of aligned segments in blocks, can be compared to the Dayhoff matrices based on percent accepted mutation (PAM) using the measure of average information per residue pair in bits units called relative entropy.
Comparison between BLOSUM 62 and PAM 160 • The BLOSUM 62 is less tolerant to substitutions involving hydrophilic amino acids, while it is more tolerant to substitutions involving hydrophobic amino acids. • For rare amino acids especially cysteine and tryptophane, BLOSUM 62 is typically more tolerant to mismatches than is PAM 160.
PAM vs BLOSUM • Dayhoff estimated mutation rates from substitutions observed in closely related proteins and extrapolated those rates to models distant relationships. • In BLOSUM approach, frequencies were obtained directly from relationships represented in the block, regardless of evolutionary distance. • The Dayhoff frequency table included 36 pairs in which no accepted point mutations.
Differences Between the PAM and BLOSUM Approach • In contrast, the pairs counted with BLOSUM, included no fewer than 2369 occurrences of any particular substitution. • The BLOSUM matrices depend only on the identity and composition of groups protein in Prosite. • Therefore, there is no expectation that these substitution matrices will change significantly in the future.
PAM Versus BLOSUM • PAM is based on an evolutionary model. • BLOSUM is based on protein families. • PAM is based on global alignment. • BLOSUM is based on local alignment.