Download

1 / 42
420 likes | 499 Views
Functional Organization of the Yeast Proteosome by Systematic Analysis of Protein Complexes.

E N D
Functional Organization of the Yeast Proteosome by Systematic Analysis of Protein Complexes Anne-Claude Gavin, Markus Bösche, Roland Krause, Paola Grandi, Martina Marzioch, Andreas Bauer, Jörg Schultz, Jens M. Rick, Anne-Marie Michon, Cristina-Maria Cruciat, Marita Remor, Christian Höfert, Malgorzata Schelder, Miro Brajenovic, Heinz Ruffner, Alejandro Merino, Karin Klein, Manuela Hudak, David Dickson, Tatjana Rudi, Volker Gnau, Angela Bauch, Sonja Bastuck, Bettina Huhse, Christina Leutwein, Marie-Anne Heurtier, Richard R. Copley, Angela Edelmann, Erich Querfurth, Vladimir Rybin, Gerard Drewes, Manfred Raida, Tewis Bouwmeester, Peer Bork, Bertrand Seraphin, Bernhard Kuster, Gitte Neubauer and Giulio Superti-Furga. Nature.Vol 415. 10 January 2002.
What is Proteomics? • Proteomics is the large-scale study of proteins, usually by biochemical methods.
Why proteomics? • “Possessing a complete genome is not sufficient to elucidate biological function” Pandy 837 • There is no strict linear relationship between genes and the protein compliment or ‘proteome’ of a cell. Ex) the presence of an ORF does not necessarily confer the existence of a functional gene.
Some possible uses of proteomics: • Protein identification • Detection of Post-translational modifications • Determination of Protein function • Molecular Medicine
Methods utilized in paper which allowed for eventual protein/protein complex identification: *Tandem Affinity Purification (TAP) *Matrix-assisted laser desorption/ ionization -time-of-flight mass spectrometry (MALDI-TOF MS).
Tandem Affinity Purification (TAP) • A procedure used to rapidly purify proteins expressed at their natural level under native conditions. • The TAP tag can be used to elute complexes from a small number of cells without prior knowledge of the complex composition, activity or function. • Can be used to identify ligands that interact with target protein.
Allows for the retention of the natural promoter so that normal levels of protein are present. Can be used to purify complexes involved in many cellular functions Can be used to study the effects of mutation on protein association of complex assembly
How TAP works *TAP tag consists of a fusion protein with two IgG binding domains: that of the Staphylococcus aureus protein A (ProtA) and a calmodulin binding peptide(CBP) separated by a Tobacco Etch Virus (TEV) cleavage site. *In the first round of elution, the sample is run through an affinity column containing IgG matrix-bound beads. Protein A binds tightly to the IgG antibody, capturing all molecules of interest that contain protein A. After washing, TEV protease is introduced to cut the protein at the TEV site.
*Eluate of this affinity column is incubated with calmodulin-coated beads in the presence of calcium. The CBP present on the eluate binds to the calmodulin beads.This column is washed to remove contaminants and remnant TEV protease. *EGTA is introduced to the recaptured complexes. The EGTA binds to the calcium, thereby releasing the purified product.
Possible Applications of TAP • Use TAP to identify other proteins which interact with the target protein. This was one of the main techniques used in this paper. (Also,reverse purification). • Use TAP to analyze structure/ activity of a purified complex • Use TAP to purify recombinant proteins.
Fig. 2. Simplified schematic of FLAG- and TAP-tag purification. (b) TAP-tag purification is a two-step purification. In the first step ProteinA is bound to IgG-Sepharose; the recombinant protein is eluted by cleavage of a sequence between the tags with TEV protease. The tagged protein is then re-purified on calmodulin–Sepharose, and eluted using a divalent metal chelator (EGTA). Tryptic digestion is performed directly in this elution buffer. The upstream sample preparation, as well as the analysis of the tryptic digests, is identical for both the FLAG and TAP.
Variations of the TAP tag • The N-Terminal Tag • The Split Tag • The Subtraction Method
The N-Terminal Tag • In rare cases, c -terminal tags impair protein function. • N- Terminal tag contains same components, just in reverse order. • Requires a promoter switching method so that the TAP tag can fuse to N-terminus while keeping this region under control of the natural promoter.
The Split Tag *The TAP tag is divided, and each half is independently fused to a different protein of the same complex. (ProtA+TEV, and CBP) *This method will allow the small quantity of complexes in which these proteins are associated to be extracted from a sample where a large percent of the proteins are unbound, or bound to other complexes. *Allows the percentage of association in vivo to be determined because TAP tag leaves proteins at natural levels.
The Split TAP tag Strategy Fig. 6 Puig et al.
The Subtraction Method Use this method when there are two or more complexes which share a common subunit, but only one of the complexes is of interest. The protein of interest is fused with normal TAP tag, protein not of interest is fused with ProtA lacking TEV site. This undesired protein is retained at IgG beads during first elution, while complex containing protein of interest is fused and proceeds on in purification process.
The Subtraction Strategy Fig. 7 Puig et al.
Background on Mass Spectrometry *Mass spec. measures the intrinsic property (molecular weight) of molecules with high sensitivity and accuracy- it is so sensitive that it can usually detect the change in mass of a peptide due to a single amino acid substitution. *The mass spectrometer determines the molecular weight of chemical compounds by generating, separating and detecting molecular ions according to mass-to-charge (m/z) ratio.
More on Mass Spec. *After the purified proteins have been run through a SDS -polyacrylamide gel with the acrylamide concentration ranging from 4% to 25% (top to bottom),the gel is either Coomassie or Silver stained. *The bands can be analyzed by mass spec., OR Mass spec can be performed on eluate without previously running it through a gel. *One benefit of running the purified products through a gel before performing mass spec. is that the gel bands can visually provide approximate stoichiometry of the fractions of peptides present (based on concentration/intensity).
Mass spec. relies on digestion of gel-separated protein bands into peptides by a sequence-specific protease (in this case, trypsin). • Work with peptides instead of whole proteins because: proteins are more difficult to elute from gel, analyze by mass spec., and molecular weight of protein isn’t sufficient marker for identification from a database. Peptides are easily eluted and provide enough information for successful identification. • There are several types of mass spectrometry- this experiment focused on MALDI/MALDI-TOF and MS/MS.
Tandem Mass. Spec. (MS/MS) • MS/MS consists of: an ion source, the first mass analyzer, the gas-phase collision cell, the second mass analyzer, and an ion detector. • The first mass analyzer is used to resolve the peptides in the mixture and isolate one species at a time (MALDI). • These isolated species are sent on to the collision cell, where they are further fragmented by collisions with inert gas.
*collision induced dissociation (CID) of the peptide precursor results in ion fragmentation that “occurs predictably at each peptide amide bond along peptide backbone, yielding a distribution of product ions in a complimentary ion series forming a ladder which is indicative of peptide sequence” (Proteomics,Timothy Palzkill) *The mass of these fragments are determined in the second mass analyzer to yield amino acid sequence. *This information can be used to search nucleic acid/protein databases. *SEE HANDOUTS.
Cont. *Advantage of (MS/MS) over MALDI fingerprinting is that sequence information derived from several peptides is more specific for identification than a list of peptide masses is.
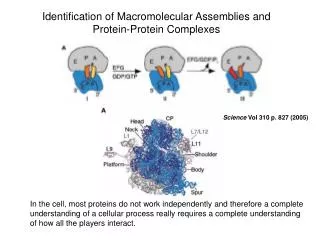
Fig. 3. Identification of interactors using AP-MS. (a) A protein of interest (along with its binding partners) is affinity-purified from biological samples and is digested with a sequence specific protease such as trypsin. (In an optional step, the protein complex may be fractionated by SDS–PAGE, and in-gel tryptic digest may be performed). The peptide mixture is then separated by liquid chromatography (reversed-phase HPLC). As peptides elute from the reversed-phase capillary column, they are ionized by electrospray and enter the mass spectrometer for analysis. (b) Within the mass spectrometer, a survey scan of the masses of individual peptides is first performed. A selected peptide (usually selected based on abundance in the survey scan) is retained for fragmentation by collision gas, generating a collision induced dissociation (CID) spectrum. Acquired CID spectra are compared with in silico generated protein databases to identify the peptide and ultimately map it to its cognate protein.
GOALS OF EXPERIMENT To use the methods of TAP and MS/MS to effectively identify novel functions of protein, and additionally to identify previously known complexes of protein assembly.
Figure 1 a. Schematic representation of the gene targeting procedure. The TAP cassette is inserted at the C terminus of a given yeast ORF by homologous recombination, generating the TAP-tagged fusion protein. b. Examples of TAP complexes purified from different subcellular compartments separated on denaturing protein gels and stained with Coomassie. Tagged proteins are indicated at the bottom. ER, endoplasmic reticulum. c. Schematic representation of the sequential steps used for the purification and identification of TAP complexes (left), and the number of experiments and success rate at each step of the procedure (right).
METHODS • Processed 1,739 genes, including 1,143 genes representing eukaryotic orthologues.(Orthologues are genes that are thought to have evolved by vertical descent from a common ancestor and are believed to have the same function).
METHODS 2 • Generated a library of 1,548 yeast strains. 1,167 strains expressed the TAP tagged protein to detectable level. • Grew cells to mid-log phase, then TAP used on total cellular lysates.
Of the589 purified TAP tagged proteins, 78% presented associated partners, showing the efficiency of this method for large scale retrieval. Possible reasons for inability to purify remaining 22% proteins: • 1)Particular proteins may not form sufficiently stable complexes. • 2)The 20K TAP tag may interfere with complex assembly or protein localization/function • 3)TAP may fail to detect transient interactions or low stoichiometric complexes • 4)Size distribution of identified proteins reveals clear bias against proteins below 15K.
Some Findings • 245 purifications corresponded to 98 known non-redundant multi-protein complexes present in the yeast protein database. • A further 242 purifications were assembled into 134 new complexes • Of all 232 TAP complexes, only 9% had no novel element. Size of TAP complexes varied from 2 to 83 components, with an average of 12 components per complex.
Figure 2 Numbers inside pie charts represent the percentages of total proteins (a) and complexes (b–f). Outer labels show partitioning of the data according to the chart function.
How function is assigned • TAP tagging of a known protein reveals the purification of several proteins with previously unknown function. Identification via mass spec. allows for novel function assignment to various proteins. • Validity of interactors is verified through reverse purification.(If A and B are associated with a TAP-tagged C, then A and C should be associated with a TAP-tagged B).
Figure 3 Primary validation of complex composition by ‘reverse’ purification: the polyadenylation machinery a, A similar band pattern is observed when different components of the polyadenylation machinery complex are used as entry points for affinity purification. Underlined are new components of the polyadenylation machinery complex for which a physical association has not yet been described. The bands of the tagged proteins are indicated by arrowheads. b, Proposed model of the polyadenylation machinery.
Figure 4 Links were established between complexes sharing at least one protein. For clarity, proteins found in more than nine complexes were omitted. The graphs were generated automatically by a relaxation algorithm that finds a local minimum in the distribution of nodes by minimizing the distance of connected nodes and maximizing distance of unconnected nodes. In the upper panel, cellular roles of the individual complexes are colour coded: red, cell cycle; dark green, signalling; dark blue, transcription, DNA maintenance, chromatin structure;pink, protein and RNA transport;orange, RNA metabolism; light green, protein synthesis and turnover; brown, cell polarity and structure; violet, intermediate and energy metabolism; light blue, membrane biogenesis and traffic. The lower panel is an example of a complex (yeast TAP-C212) linked to two other complexes (yeast TAP-C77 and TAP-C110) by shared components. It illustrates the connection between the protein and complexlevels of organization. Red lines indicate physical interactions as listed in YPD22.
More Findings • Orthologous proteins preferentially interact with complexes enriched with other orthologues. • Nonorthologous proteins did not display such an interaction. • Rate of interaction with essential gene products is greater for essential than for nonessential proteins.
Figure 5 Comparison of three TAP protein complexes isolated from human and yeast cells. All orthologous pairs are indicated by arrows, demonstrating that the complex composition between yeast and human is largely conserved. Coomassie-stained gels are shown only for the human purifications. a, Arp2/3 complex; b, Ccr4–Not2 complex; c, Trapp complex. Hyp. protein, hypothetical protein.
TAP to locate Orthologues *Via TAP of human NOT2 and yeast Ccr4, the authors were able to determine that the human and yeast Ccr4-Not complexes are comparable in subunit composition *TAP of Arp2 in yeast and ARPC2 in humans *Purified and characterized an orthologous human TRAPP (transport protein particle). **THESE EXAMPLES SHOW THAT YEAST ANAYLSIS CAN OFTEN PREDICT COMPOSITION OF HUMAN ORTHOLOGUE.
FURTHER STUDIES • Evaluation of the impact of systematic modification of experimental parameters on complex integrity. • Study of the concept of a eukaryotic ‘core proteome’
Figure 2 (A) Plasmid maps including the C-and N-terminal TAP tagging cassettes. Single stars indicate TEV protease cleavage sites; two stars indicate the enterokinase cleavage site. (B) Structure of the oligonucleotides used for tagging.
Fig. 4. N-Terminal tagging strategy. A PCR fragment, is amplified included the tagging cassette and flanking regions of homology to the target gene.Following transformation into yeast cells, the PCR fragment integrates into the genome, placing the target ORF under control of the GAL1 promoter. In the final step, Cre recombinase is used to remove the marker and the GAL1 promoter, leaving the N-terminal TAP-tagged ORF under the control of its natural promoter.
The Tandem Affinity Purification (TAP) Method: A General Procedure of Protein Complex Purification Oscar Puig1, Friederike Caspary, Guillaume Rigaut, Berthold Rutz, Emmanuelle Bouveret, Elisabeth Bragado-Nilsson, Matthias Wilm and Bertrand Séraphin European Molecular Biology Laboratory Meyerhofstrasse, Heidelberg, D-69117, Germany Available online 22 February 2002.
Affinity-purification mass spectrometry (AP-MS) of serine/threonine phosphatases Ginny I. Chena and Anne-Claude Samuel Lunenfeld Research Institute at Mount Sinai Hospital, Department of Medical Genetics and Microbiology and Graduate Department of Molecular and Medical Genetics, University of Toronto, 600 University Avenue, Room 992A, Toronto, ON, Canada M5G 1X5 Accepted 15 February 2007. Available online 29 May 2007.