Download

1 / 34
370 likes | 803 Views
Clash Of The Titans! Convergence Of Tau And Alpha-Synuclein Amyloid In Neurodegenerative Diseases. John Q. Trojanowski, M.D., Ph.D. Institute on Aging Center for Neurodegenerative Disease Research Department of Pathology and Laboratory Medicine University of Pennsylvania Philadelphia, PA.

E N D
Clash Of The Titans! Convergence Of Tau And Alpha-Synuclein Amyloid In Neurodegenerative Diseases John Q. Trojanowski, M.D., Ph.D. Institute on Aging Center for Neurodegenerative Disease Research Department of Pathology and Laboratory Medicine University of Pennsylvania Philadelphia, PA
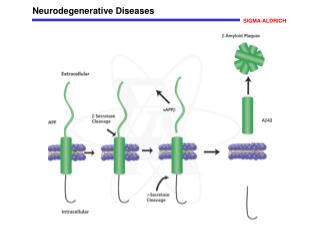
Neurodegenerative Diseases Characterized by Brain Amyloidosis Disease LesionsComponents Parkinson’s Disease LBs -Synuclein Dementia with Lewy Bodies LBs -Synuclein Multiple System Atrophy GCIs -Synuclein Alzheimer’s Disease SPs Aβ (Most common synucleinopathy!)NFTs Tau LBs α-Synuclein Prion diseases SPs Prions Tauopathies NFTs Tau Trinucleotide Inclusions Expanded Repeat Expansion PolyQ tracts
US Population Demographics 85 Years of Age or Older: 1950-2050 High Median Millions Low Year
Synucleinopathies • Parkinson’s disease - familial and sporadic* • Dementia with Lewy bodies • Multiple system atrophy • Neurodegeneration with brain iron accumulation-1 (formerly H-SD) • Pure autonomic failure • REM sleep behavior disorder • Down syndrome* • Alzheimer’s disease* (The most common α-synuclein brain amyloidosis!) • * These disorders are “triple” brain amyloidoses with tau, Aβ and α-synuclein amyloid deposits
Normal Alpha Synuclein • An abundant low Mr synaptic protein, present to a lesser extent in perikarya and axons, but also in oligodendroglia • Other members of the synuclein family of synaptic proteins include beta- and gamma-synuclein • Function is unknown but may play roles in synaptic transmission • Is a phosphoprotein, but role of alpha-synuclein phosphorylation in its normal function is unknown
Alpha-Synuclein Mutations Cause Familial Parkinson’s Disease = 6 imperfect repeats of 11 amino acids with the conserved core KTKEGV NAC peptide (aa 61-95) A30P mutation A53T mutation H2N COOH Negatively Charged Hydrophobic Middle Carboxy-terminus Section Mutations promote protein aggregation and filament formation
Pathological Alpha-Synuclein • Forms insoluble filamentous aggregates with the properties of amyloid • Amino acids 71-82 in the NAC domain are the minimal, essential sequences required for fibrilization • Filamentous alpha-synuclein inclusions form in neuronal perikarya, processes and in gliaI cells • Is abnormally phosphorylated, nitrated and ubiquitinated
Alpha-Synuclein Pathological Inclusions Glial cytoplasmic inclusions Lewy neurites Lewy bodies Lewy bodies Electon microscopy Glial cytoplasmic inclusions Electron microscopy
Environmental and/or Genetic Risk Factors ? Alpha-synuclein Dysfunction And Aggregation Play Central Roles in Mechanisms of Neurodegenerative Disease Genetic Factors -Synuclein Mutations or Duplications APP, PS1, PS2 Mutations, DS Synuclein Dysfunction and/or Aggregation Neurodegeneration
But hold on! Tau and Alpha-Synuclein Pathology Commonly Co-occurr in Neurodegenerative Diseases – So what does this mean? • Diseases with tau, α-synuclein and abundant Ab deposits • Lewy body variant of Alzheimer’s disease • Alzheimer’s disease • Familial Alzheimer’s disease • Down’s syndrome • Diseases with tau, α-synuclein and scant/no Ab deposits • Parkinson’s disease/Dementia with Lewy bodies • Multiple system atrophy • Guam ALS/PDC • Neurodegeneration with brain iron accumulation type 1 • Some cases of PSP, CBD, and Pick’s disease
Normal Tau Proteins • Abundant low molecular weight microtubule (MT) associated proteins localized mainly in axons • Promote MT polymerization, bind to MTs and stabilize MTs in the polymerized state • Normally is phosphorylated at a range of Ser and Thr residues • Phosphorylation negatively regulates binding of tau to MTs
4R2N 3R2N 4R1N 3R1N 4R0N 3R0N Six Human Brain Tau Isoforms Are Generated By Alternative Splicing SDS-PAGE E2 E3 E10 441 1 R1 R2 R3 R4 67kDa 410 1 R1 R3 R4 62kDa 412 1 59kDa R1 R2 R3 R4 381 54kDa 1 R3 R4 R1 52kDa 383 1 R1 R3 R4 R2 48kDa 352 1 R1 R3 R4 R2
PHF-Tau Proteins • Insoluble • Form filamentous amyloid deposits in neuronal cell bodies/processes and glia • Aberrantly hyperphosphorylated at Ser/Thr; ubiquitinated • Unable to bind to MTs unless dephosphorylated in vitro
Tau Mutations Cause FTDP-17 By Different Mechanisms And Aggregation Of Tau Disrupts Axonal Transport Leading to Neurodegeneration S305N/S N279K P301L/S L284L V337M E342V G389R K257T D280K I260V G272V R406W 1 441 Exon 10 +3 N279K D280K L284L P301S P301L S305N S305S C A T U C G DAAG a g u Intron 9 a AAUAAGAAGCUGGAUCUU----------CCGGGAGGCGGCAG-U g u g a g c a c a c u c u u c u/t u g t Intron 10 +16 +14 +13 +12 Mutations Altering Exon 10 Splicing -N279K -D280K -L284L -S305N -S305S -Intron 10 mutations Mutations Promote Tau Aggregation -G272V -P301L –P301S -V337M Mutations Impairing Tau Protein Function -G272V -D280K -P301L -P301S -V337M -G389R -R406W
Tau Positive Inclusions in Neurons AD GUAM CBD Pick’s disease FTDP17 DEM PEG PSP
Paired Helical Filaments (PHFs) Formed By Tau Are Building Blocks Of NFTs In AD Brain • Two twisting strands with an apparent periodicity of 80nm and an alternating width between 8 and 20nm • Molecular composition tau From Lee et al. Science (1991) 251, 675-8
72kDa 68kDa 2N4R 67kDa 64kDa 2N3R 62kDa 60kDa 1N4R 59kDa 1N3R 54kDa 52kDa 0N4R 0N3R 48kDa Sarkosyl-insoluble Tau Bands Before and After Dephosphorylation - + - + - + Dephos. • AD • ALS/PDC • Down’s syndrome • FTDP-17 (G272V, V337M,etc.) • GSS • Nieman-Pick disease type C • FTDP-17 (DK280) • Pick’s disease • CBD • FTDP-17 (mutations in I10, L284L, etc.) • PSP
Tau Dysfunction And The Pathogenesis Of AD And Related Neurodegenerative Tauopathies Genetic Factors Environmental Factors Tau Mutations APP, PS1, PS2 Mutations ? ? Hyperphosphorylation Perturbation of 4R/3R Ratio Loss of Tau Function Gain of Toxic Function Kinases DeP-Tau P-Tau Phophotases Tau Dysfunction Tau Aggregation/ MT Loss Impaired Transport & Neurodegeneration
Tau Pathology in Patients with the A53T Alpha-Synuclein Mutation a-syn Tau
Summary of -Synuclein Assembly Studies • -Synuclein readily assembles into 10 nm diameter filaments • A53T mutation facilitates -synuclein assembly • -Synuclein is incapable of fibrillogenesis • Residue 71-82 is required for -synuclein filament assembly
HSP70 PROTECTS AGAINST ALPHA- SYNUCLEIN INDUCED DOPAMINERGIC NEURON DEGENERTATION (Auluck et al., Science 2002)
Characterization of -Synuclein Transgenic Mice (Giasson et al., Neuron, 2002)
Abundant -synuclein Inclusions in A53T -synuclein Transgenic Mice Syn 303 SNL-4 SNL-4 SNL-4 Spinal cord Spinal cord Spinal cord Spinal cord Syn 505 Syn 303 Syn 303 Syn 505 pons pons midbrain Raphe Syn 506 Syn 303 Syn 505 Syn 505 pons locus ceruleus pons cerebellum
Accumulation of Detergent Insoluble -synuclein in the Spinal Cord of A53T Tg Mice
α-Synuclein Filaments can be Isolated from A53T Transgenic Mice
Tau Pathology in the Spinal Cord and Midbrain of A53T a-Syn Tg Mice tau a-syn tau tau tau overlay tau a-syn
Lessons From Nicoll, et al. (Nat. Med., 2003) For Therapy Of Brain Amyloidoses • Subtraction of Aβfrom the brains of patients with diseases having abundant tau, alpha-synuclein & Ab pathologies • Lewy body variant of Alzheimer’s disease • Sporadic/Familial AD • Down’s syndrome • Will convert them to phenocopies of diseases with abundant tau and/or alpha-synuclein, but scant/no Ab pathologies • Parkinson’s disease/Dementia with Lewy bodies • Guam Marianna dementia/PDC
^ ^ ^ Model of Fibrilization in Neurodegenerative Disease = -helix = random coil = -pleated sheet
The Deleterious Consequences Of Protein Misfolding N N C C Inclusions Cell Death mRNA Ribo- somes chaperone N N C Misfolded Small oligomers N Cell Death Proteasome
More Deleterious Consequences Of Protein Misfolding Microtubules a-synuclein tau vesicles Improper trafficking Increased abundance Tau hyperphosphorylation Pathological aggregates
Mark Forman Benoit Giasson Makoto Higuchi Hiro Uryu Bin Zhang CNDR Members Virginia Lee N. Bonini, J. Duda J. Galvin, D. Galasko L. Golbe, M. Grossman H. Hurtig, T. Iwatsubo C. Lippa, B. Miller D. Murphy, M. Stern Nat’l AD Coordinating Center NIA Alzheimer Disease Centers Penn Head Injury Center It Takes Great A Team! Supported by Grants from the NIA, the Alzheimer’s Association, Michael J. Fox Foundation and the Families of our Patients
DISCLAIMER If you attended to the content of this lecture, you may have participated in effortful mental activity, and this may reduce your risk for dementia. * However, this lecture is not intended as a therapeutic intervention and there is no guarantee that this lecture has therapeutic benefit. * Wilson et al. JAMA 287:742-748, 2003; Verghese et al. NEJM, 34:2508-2516, 2003