Download
1 / 16
160 likes | 337 Views
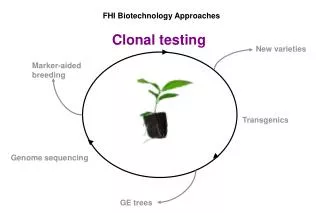
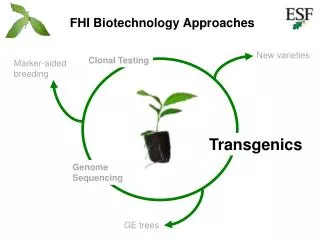
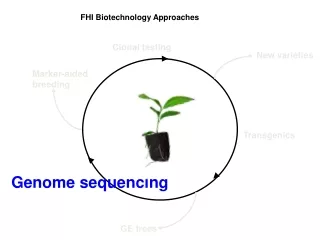
FHI Biotechnology Approaches. Clonal testing. New varieties. Marker-aided breeding. Transgenics. Genome sequencing. GE trees. Chestnut Genome Research Team. John E. Carlson, PI, Schatz Center, Penn State University. DNA Sequencing Stephan C. Schuster

E N D
FHI Biotechnology Approaches Clonal testing New varieties Marker-aided breeding Transgenics Genome sequencing GE trees
Chestnut Genome Research Team John E. Carlson, PI, Schatz Center, Penn State University DNA Sequencing Stephan C. Schuster Professor of Biochemistry and Molecular Biology, Penn State Lynn P. Tomsho, Daniela Drautz, and Lindsay Kasson Sequencing Specialists, Penn State Tyler Wagner Research Assistant, Penn State Bioinformatics and Comparative Genomics Webb Miller Professor of Biology and Computer Science & Engineering, Penn State Charles Addo-Quaye Postdoctoral Fellow, Penn State Meg Staton, Stephen Ficklin and Christopher Saski Bioinformatics team at Clemson University Genomics Institute Abdelali Barakat Research Associate, Clemson University FHI Cooperators: Bert Abbott, Sandra Anagnostakis, Kathleen Baier, Ali Barakat, Nurul Faridi, Eric Feng, Stephen Ficklin, Fred Hebard, Thomas Kubisiak, Charles Maynard, Scott Merkle, Joseph Nairn, William Powell, Dana Nelson
The Chinese Chestnut Genome Sequencing Project Our Goals: 1) Develop a complete reference genome sequence for chestnut 2) Identify all genes in the three blight resistance QTL 3) Deliver candidate genes to the FHI Transgenics group and the FHI Marker-aided breedinggroup 4) Provide the genome to the research community 5) Demonstrate the potential of genomics to address forest health and ecosystem restoration.
The Chinese Chestnut Genome Sequencing Project DELIVERABLES FOR YEAR ONE were all achieved The reference Castanea mollissima cv. Vanuxem genome was sequenced to over 25-fold depth. Preliminary de novo assemblies of the reference genome sequence were conducted. Commenced use of genetic and physical map information (from the FHI genetic technologies group) in genome assembly.
The Chinese Chestnut Genome Sequencing Project DELIVERABLES FOR YEAR ONE, the details • “Shot-gun” sequencing completed by March, 2010 • 18-fold* depth by 454 technology = 14.2 Gigabases • 47-fold* depth by Illumina technology = 37.6 Gigabases • Passed QC tests: • mtDNA < 0.4% and cpDNA < 0.3% of sequence • microbial DNA negligible • sequence reads over 350 bp • repetitive DNA manageable (conserved repeats at 9 to 12%) • Preliminary assemblies of the genome sequence were promising totalling app. 852 Mbp, but in smaller pieces than desired • * assumes a genome size for chestnut of app 800 Mbp
The Chinese Chestnut Genome Sequencing Project WHAT WE LEARNED IN YEAR ONE “Next Gen” sequencing technologies produce a large amount of high quality data, very quickly. Large amounts of high quality data take a long time to assemble using currently available software. Assembly of the reference genome will require more than just “shot gun” Next Gen sequence data. “Paired end” data are required to pull contigs together into chromosome scaffolds. For assembly purposes, the chestnut genome may be larger than 800 Mbp.
The Chinese Chestnut Genome Sequencing Project DELIVERABLES ACHIEVED IN YEAR TWO • Produced paired-end sequence data. • Covered the physical map with BAC-end sequences. • Commenced gene identification and characterization: • Transcripts aligned to the genome assembly • Assembly searched for genes • Preliminary annotations of genes conducted • Strategy for resistance gene discovery updated.
The Chinese Chestnut Genome Sequencing Project DELIVERABLES FOR YEAR TWO, the details • Paired-end sequences from 454 sequencing at 4.5-fold depth (3.6 Gb). • 43,143 BAC-end sequences obtained, “tiling” the physical genome map to 1.5-fold depth, anchored to genetic map. • New assemblies conducted using the paired-end data: • 587,208,063 bp assembled into 51,766 scaffolds, • 925,312,071 bp assembled into 1,147,939 contigs
The Chinese Chestnut Genome Sequencing Project DELIVERABLES FOR YEAR TWO Gene Identification and Characterization • Chinese chestnut unigenes (transcripts) from NSF project aligned well to the current genome assembly: • 97% of transcripts (46,954) aligned to genome assembly • 98% identity of transcripts and genome sequences • Results of gene search with preliminary assembly: • 66,662 gene models predicted in the scaffolds • - certainly an over-estimate of gene number at this point • - mean gene length 2,761 bp, maximum length 43,203 bp • - mean number of genes per scaffold 12.8, maximum 58 • Candidate gene sequences identified in genome contigs • Coding sequences delivered to the transgenics team
The Chinese Chestnut Genome Sequencing Project The largest gene identified in the preliminary Chinese Chestnut genome assembly Homolog of AT1G67120 (NP_176883.4), AAA ATPase, von Willebrand factor type A domain-containing protein, with nucleoside-triphosphatase activity. • Transcript length: 43,203 bases • Number of Exons: 71 • Scaffold ID: scaffold01252
The Chinese Chestnut Genome Sequencing Project Most Arabidopsis single-copy genes have strong matches to the current genome assembly (by BLAST alignment) N = 959 Number of Genes E-values (strength of matches)
The Chinese Chestnut Genome Sequencing Project Best matches of proteins from the chestnut genome assembly are to peach and other related species Only 1% of best matches to Arabidopsis. The peach genome is best for chestnut gene discovery. Best matches: • peach, 23% • rice, 12% • grapevine, 7% • Eurosids 1 species, 56% BLASTx alignments to model plant genomes in Phytozome
The Chinese Chestnut Genome Sequencing Project The predicted chestnut proteins are most similar to species in the Eurosids 1 clade, that also includes peach and chestnut. eurosids 1 eurosids 2 Source: http://www.phytozome.net/
The Chinese Chestnut Genome Sequencing Project However, the genome assembly is uneven and not as good as needed to assemble all of the blight resistance QTL genes Range of coverage among genome scaffolds
The Chinese Chestnut Genome Sequencing Project Our target is the blight resistance genes. We will sequence the Resistance QTL themselves, which is already in progress: • Sets of BAC contigs covering the QTLs were identified. • Sequencing of each QTL underway as contig pools. • Genes will be identified using peach resistance QTL and CC transcripts.
The Chinese Chestnut Genome Sequencing Project Clonal testing Year 3 - Gene discovery Marker-aided breeding New varieties Markers in QTL genes Transgenics Genome sequencing Candidate gene validation Complete QTL sequences Candidate genes from the QTLs GE trees