Download

1 / 39

E N D
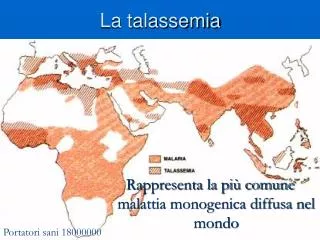
La Talassemia E’ una malattia ereditaria del sangue, a trasmissione autosomica recessiva, con alta prevalenza in alcune aree del mondo, in particolare bacino del Mediterraneo (Italia, Grecia, Turchia, Cipro, Marocco, Arabia saudita) e nel Sud-Est asiatico (India, Vietnam, Cambogia, Giappone). Essa è causata da una sintesi assente o deficitaria di una o più catene polipeptidiche della globina. Ne esistono due forme principali, talassemia α e talassemia β, a seconda che la mutazione interessi il gene per la catena α o quello per la catena β. La forma più frequente in Italia è la β- talassemia
Talassemia Classificazione clinica Si distinguono tre condizioni di gravità crescente: • Portatore asintomatico • Talassemia intermedia • Talassemia major
Talassemia Portatore asintomatico: Clinicamente silente, è caratterizzato: • aumento dei G.R • microcitosi con riduzione di MCV(60-70 fl) • Ipocromia con riduzione di MCH (19-23 pg) • Alterazioni morfologiche degli eritrociti (anisopoichilocitosi) • Aumento delle resistenze globulari osmotiche • L’elettroforesi dell’Hb dimostra aumento dell’HbA2 (4-6%) • nel 30% dei portatori si ha anche un modesto aumento dell’Hb F(2-5%). TERAPIA: nessuna
Talassemia Talassemia intermedia: Comprende un gruppo eterogeneo di pazienti con genotipo e gravità clinica variabile (da casi con minime alterazioni ad altri con manifestazioni simili a quelli della t. major). • Pattern emoglobinico variabile a seconda del genotipo ma comunque sempre con aumento della HbF (in genere >15-20%) • A causa dell’eritropoiesi inefficace si accumula ferro nell’organismo. TERAPIA: • Ferrochelanti (desferoxamina s.c.); • Acido folico (per evitare deficit da iperconsumo); • Splenectomia (dopo il 4°-5° anno). Non necessita di trasfusioni per la sopravvivenza
Hb GR Talassemia major La Talassemia major è caratterizzata da sintesi ridottissima o del tutto assente di catene β-globiniche e quindi di HbA, con conseguentegrave anemia. Le catene in eccesso si legano alle catene γ determinando la formazione di HbF che, rispetto all’HbA, ha una maggiore affinità per l’O2 e lo cede con maggiore difficoltà ai tessuti.
Quali sono le conseguenze della Talassemia? • Inadeguata produzione delle catene globiniche • ipocromia e microcitosi • accumulo non bilanciato di altre catene globiniche • precipitati negli eritroblasti • Ciò determina: • eritropoiesi inefficace • ridotto numero di eritrociti maturi • distruzione periferica con anemia emolitica
Talassemia major Insorgenza: (4-6 mesi di età) Sintomi clinici alla presentazione: • Pallore, colorito itterico • Febbricola • Irritabilità • Turbe dell’alvo • Episodi infettivi ricorrenti • Splenomegalia (per iperplasia della polpa rossa secondaria all’esaltata eritrocateresi) • Epatomegalia (per persistenza post fetale di mielopoiesi extramidollare e per alterazioni del circolo epato-splenico)
Talassemia major QUADRO EMATOLOGICO • Hb 4-6 g/dl • Globuli rossi 2.000.000/mmc • Presenza di eritroblasti ortocromatici nel sangue periferico • Anisopoichilocitosi, cellule a bersaglio • Leucociti e piastrine normali o ridotti per sequestro splenico • Resistenza globulari aumentate • Iperplasia eritroblastica midollare • Hb F: 70-90% • Hb A2 > 3.5%
Talassemia Major striscio di sangue periferico
Talassemia major ALTERAZIONI OSSEE • Secondarie all’iperplasia del tessuto emopoietico nelle ossa spugnose • Alterazioni caratteristiche: facies simil-asiatica • Alterazioni radiologiche caratteristiche (per es. cranio a spazzola, osteoporosi) • Deformità articolari e prematura fusione delle epifisi
Talassemia major Deformazioni faciali, facies simil-asiatica
Talassemia major Rarefazione delle corticali ossee, cranio a spazzola
Talassemia major Difettiva chiusura delle epifisi delle mani e delle teste omerali
Talassemia major SOVRACCARICO DI FERRO Per: • Aumentato assorbimento di ferro • Carico emotrasfusionale I sintomi del sovraccarico di ferro si sviluppano soprattutto con l’età. • Organi bersaglio: fegato, cuore, ghiandole endocrine (pancreas, paratiroidi, gonadi, ipofisi, tiroide) • Quadri clinici più invalidanti: insufficienza cardiaca, diabete, cirrosi epatica
TERAPIA DELLA TALASSEMIA MAJOR • Terapia trasfusionale (target: Hb > 10.5-11 g/dL) • Terapia chelante del ferro • Splenectomia • Trapianto di cellule staminali emopoietiche allogeniche
Terapia Trasfusionale Correzione anemia Soppressione eccesso di eritropoiesi Inibizione assorbimento intestinale di Fe legato all’anemia Talassemia Major
Terapia Ferrochelante • Desferoxamina (DFO): per via sottocutanea ad infusione lenta • Deferiprone (L1): • per via orale • ICL 670 A: • per via orale • GT56-252: • Un chelante orale in via di sperimentazione Talassemia Major
Complicanze nella talassemia major • Epatiche: epatiti acute e croniche. Emocromatosi • Cardiache: Pericarditi, Miocardioptie, Insufficienza Cardiaca • Endocrine: Ipogonadismo, Diabete mellito, Osteoporosi, Ipotiroidismo, Ipoparatiroidismo. • Emolisi:Alloimmunizzazione anti eritrocitaria, anti Rh • Infezioni
Talassemia Major OBIETTIVI DEL TRATTAMENTO Miglioramento della Qualità della vita Inserimento scolastico Attività lavorativa normale Accrescimento normale Riduzione iperplasia midollare prevenzione delle alterazioni scheletriche Riduzione della splenomegalia e del conseguente ipersplenismo Riduzione dell’ipervolemia minor sovraccarico cardiaco Istituto “Seragnoli”-Bologna
Terapia non convenzionale: • Trapianto di midollo osseo (TMO) • Trapianto di cellule staminali • Terapia genica
Sostituire il compartimento emopoietico alterato con un patrimonio di cellule staminali ottenuto da un donatore sano capace di ricostituire il sistema emopoietico ed immunitario del ricevente Terapia non convenzionale: OBIETTIVO GUARIGIONE
Fonti di cellule staminali • Midollo osseo • Sangue periferico • Cordone ombelicale • Tessuti fetali • Embrioni
Trapianto di midollo osseo • I geni determinanti la compatibilità tissutale sono raggruppati come Complesso Maggiore di Istocompatibilità (MHC) – HLA • Requisito fondamentale per il TMO è la disponibilità di un donatore HLA-compatibile
TMO allogenico • Da donatore singenico (gemello monocoriale) • Da donatore familiare HLA identico • Da donatore familiare HLA non - identico • Da donatore HLA identico non - correlato
Prevenzione della talassemia Essa si fonda su: • Identificazione dei portatori (i c.d. “microcitemici”) mediante esami di screening effettuati nelle scuole o in epoca prematrimoniale. • Diagnosi prenatale
PREVENZIONE TALASSEMIA Diagnosi prenatale (per una scelta consapevole ) Screening (Per l’identificazione del portatore sano) Campagne di informazione Consulenza genetica
Screeningesami di I Livello • Esame emocromocitometrico • dosaggio dell’HbA2 (con HPLC) • valutazione qualitativa di Hb patologiche (mediante elettroforesi) • dosaggio di HbF (con HPLC) • dosaggio enzimatico della Znpp e/o dosaggio ematico della ferritina
Screeningesami di II Livello Studio dei familiari
Screeningesami di III Livello Studio del DNA del paziente e/o dei familiari per l’identificazione della mutazione
La diagnosi prenatale si basa sull'identificazione diretta di mutazioni nel DNA fetale in gravidanze a rischio. Tale indagine non prevede nessun tipo di rischio né per la madre per il feto. E’ una tappa obbligatoria per l’esecuzione del trapianto in utero La diagnosi prenatale
La diagnosi prenatale • Prelievo di sangue dai villi coriali • (Villocentesi) • (VIII- X settimana di gestazione) 2) Prelievo di sangue fetale (Cordocentesi) (XVIII- XXII settimana)
-TALASSEMIE • Diffuse prevalentemente nel Sud-Est asiatico • Quadri clinico-ematologici variabili a seconda del grado di riduzione nella produzione delle catene • Se mancano 1-2 geni : • quadro clinico sovrapponibile a talassemia minor. • Hb Bart (4) alla nascita 5-10%, Hb normale in età adulta (diagnosi di esclusione) • Se mancano 3-4 geni : • idrope fetale (80% Hb Bart): incompatibile con la vita • malattia da HbH (4). Severa anemia ipocromica, microcitica, con componente emolitica, splenomegalia. • Hbs Gower-1 (zeta 2 epsilon 2), Gower-2 (alpha 2 epsilon 2), and Portland-2 (zeta 2 beta 2).
Talassemia : ipocromia, microcitosi, cellule bersaglio e poichilocitosi Malattia da HbH: reticolociti e eritrociti con aggregati di catene ß