Download

1 / 71
710 likes | 838 Views
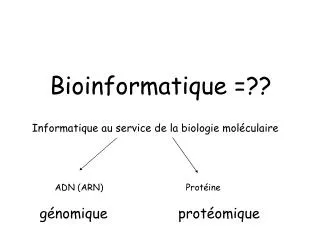
Bioinformatique ABI-1001. 2ème Partie: La Biologie Structurale Cours #2 Domaines, classification et prédictions. Domaines: définition. Les domaines peuvent être définis de différentes façons:. Séquence: hérédité de (sous)séquences. Fonction: modules fonctionnels protéiques.

E N D
BioinformatiqueABI-1001 2ème Partie: La Biologie Structurale Cours #2 Domaines, classification et prédictions
Domaines: définition • Les domaines peuvent être définis de différentes façons: • Séquence: hérédité de (sous)séquences. • Fonction: modules fonctionnels protéiques. • Structure: unité de repliement ou contacts atomiques. • Fragment de structure capable de se replier ou d’adopter une conformation indépendamment du reste de la structure.
Domaines: définition • Domaine: Biologie structurale • Région d’une prot. qui possède son propre cœur hydrophobe et qui est structurellement indépendant du reste de la prot. généralement colinéaire en séquence. Biochimie/Génétique • Fragments minimale d’un gène capable d’effectuer une fonction donnée. • Module:domaines similaires se retrouvant dans plusieurs prot. (ex. Immunoglobine, SH2)
N N N N N Domaines: évolution… N Sélection: intermédiaires ancestraux Protéines multi-domaines N Domaines ancestraux (ère pré-biotique) N Hétéro-dimères Fusion de gènes N N
Identification des domaines structuraux • Les domaines sont omniprésents dans les protéines: • La plupart des grosses protéines sont composées d’unités semi-indépendantes: • Modularité. • Efficacité de repliement. • Ségrégation fonctionnelle. D-Amino Acid Oxidase HYDROXYBENZOATE HYDROXYLASE: 1PHH FAD/NAD(P)-binding domain
Identification des domaines structuraux • > 60% des protéines ne sont constituées que d’un seul domaine. • Environ 70% des protéines multi-domaines n’ont pas plus de 2 domaines. • Il existe une relation linéaire entre la taille des protéines (en acides aminés) et le nombre de domaine qu’elles possèdent. N=787 Jones S et al. (1998) Protein Science 7:233
Identification des domaines structuraux • La grosseur moyenne des domaines se situe environ à ~ 100 résidus. • Approximativement 70% des domaines sont continus en séquence. N=787 Jones S et al. (1998) Protein Science 7:233
Identification des domaines structuraux • Quel est l’importance d’identifier ces domaines: • L’évolution s’effectue au niveau de la séquence mais sélectionne des fonctions. • L’identification des domaines dans une structure peut révéler des unités fonctionnelles qui ont évoluées indépendamment. L’alignement et la recherche de séquence en fonction des domaines est beaucoup plus spécifique. • Les domaines sont les unités fondamentales de l’analyse de la structure des protéines.
D1 D2 D1 D2 N C D1 D2 N C D1 D2 N C Identification des domaines structuraux • Concept: interactions atomiques à l’intérieur d’un domaine sont plus nombreuses qu’entre domaines. • La quête: groupes de résidus avec le max. de contact entre résidus mais avec un min. de contact avec d’autres groupes de résidus. • Problème: parfois ces groupes appartiennent à des segments non-contiguës ⇒ discontinuité séquentielle du domaine.
Identification des domaines structuraux • L’identification des domaines est en fait un problème d’optimisation. • Plusieurs méthodes ou algorithmes: • STRUDL • DomainParser • PUU • DETECTIVE • DOMAK Jones S et al. (1998) Protein Science 7:233
Identification des domaines structuraux • Comment peut-on évaluer l’efficacité d’une méthode? • Teste la méthode avec un groupe de protéines dont les domaines sont connus (experts) avec et/ou d’autres méthodes. Jones S et al. (1998) Protein Science 7:233
Identification des domaines structuraux • La méthode la plus efficace reste tout de même l’identification manuelle. • Mise à jour difficile vue le nombre croissant de structures. Jones S et al. (1998) Protein Science 7:233
Identification des domaines structuraux • CATH http://www.biochem.ucl.ac.uk/bsm/cath/ • SCOP • http://scop.berkeley.edu/ • 3Dee http://www.compbio.dundee.ac.uk/3Dee/
Classification? • La structure d’une protéine peut nous renseigner sur ses fonctions et son passé évolutif. • Nécessite la connaissance de la structure et de sa relation avec les autres protéines. • Requiert un savoir générale du repliement de la protéine et des informations détaillées sur la structure de beaucoup de protéines. • D’un point de vue évolutif, la structure est beaucoup mieux conservé que la séquence.
??? Le repliement évolutif ??? • Protéines divergentes du point de vue séquence, retiennent l’architecture et la topologie de leur repliement ancestrale. • Les prot. peuvent avoir plus d’un repliement énergétiquement stable. pression sélective = contraintes fonctionnelles. • Les similarités structurales entre prot. sans parenté s’explique souvent par la convergence d’un repliement stable plutôt que par la divergence d’un ancêtre commun. évolution parallèle. • Évolution nécessite la subsistance fonctionnel des prot. existantes sinon…R.I.P.
Structural Classification Of Proteins • Organisation hiérarchique des prot. selon leur structure et leur origine évolutive. • Procure un réductionnisme qui facilite les comparaisons structurales. • Inspection/comparaison automatique et visuel • L’unité principale de catégorisation son les domaines; ceux-ci étant l’unité évolutive, fonctionnelle et structurale des prot.
Structural Classification Of Proteins • Niveaux hiérarchiques de SCOP: • Class: dérive du contenu en structure secondaire. • Fold:provient de la topologie, l’arrangement et l’orientation des éléments de structures secondaires. (968) • Similarités structurales proviennent des propriétés physico-chimique ≠ évolution • Superfamilly: regroupent les prot. Avec peu d’identité de séquence mais dont la structure et la fonction sont relié. (~1300) • Origine évolutive commune probable • Familly: Structure et fonction similaire ainsi que > 30% d’identité de séquence (~2300) • Origine évolutive certaine
Structural Classification Of Proteins 2 domaines en tout Relié à l’activité catalytique 3 chaînes dans le fichiers pdb dont le domaine inclus les a.a. de 181 à 419 Rasmol Liens externes Chime
Structural Classification Of Proteins Swiss-PDBviewer Pour connaître le second domaine
Classification, Architecture, TopologyandHomology • Classification semi-automatique: • Selon des protocoles et algorithmes (PUU, DETECTIVE et DOMAK). • Valider manuellement en cas d’ambiguïté. • Ne considère que les structures résolu à une résolution de 3 Å ou moins. • Les prot. multi-domaines sont subdivisées selon leur domaines constitutifs de manières automatique.
Classification, Architecture, TopologyandHomology • Quatre niveaux majeurs de classification: • Class: déterminer automatiquement par la composition en structures secondaires. • Architecture:déterminer manuellement selon l’orientation des éléments de structures secondaires sans toutefois tenir compte de la connectivité. • Topology:classer automatiquement en famille de conformation (fold family) selon la forme globale et la connectivité des éléments structuraux. • Homology(superfamille):regroupe les domaines de prot. ayant un ancêtre commun (homologue). • Sequence families: regroupe à l’intérieur de superfamilles les homologues selon leur identité de séquence
Classification, Architecture, TopologyandHomology • 4 Classes: • Mainly a • Mainly b • Mixed a & b • Few secondary structures • 37 Architectures • 813 Topologies • 1467 Homologues MainlyaMainlyb Few secondary structures Mixed a & b
Classification, Architecture, TopologyandHomology http://www.biochem.ucl.ac.uk/bsm/cath/ • Le site de CATH contient d’autres ressources utiles: • SSAP server: comparaison structurale de 2 prot. et alignement structurale • GRATH server: recherche de topologie pour une structure donnée. • DHS: dictionnaire des homologues. • Gene3D: Base de données d’identification de gène structuraux pré-calculés à travers 120 génomes. • IMPALA: Comparaison de séq. avec une base de données PSI-BLAST.
CATH, exemple…1GTM 2 domaines/chaînes
CATH, exemple…1GTM Généralement relié à l’activité
Classification, Architecture, TopologyandHomology • Il semblerait que les prot. peuvent adopter environ 1000/5000 conformations différentes. (on est loin de 20300 pour une protéine moyenne: nombre qui soit dit en passant est plus grand que le nombre d’atome dans l’univers) • À ce jour on connaît au moins un représentant de chacune de ces conformations. • Environ une dizaine de topologies contiennent plus de la moitié de toutes les prot.
Comparaison et Alignement de structure • Importance de la comparaison et l’alignement: • Classification: assignation de classe de repliement et éventuellement création de bibliothèques de configuration. • Fonction: identification de la fonction d’une prot. inconnu en comparant avec la structure de prot. connue. • Prédiction: évaluation de la qualité d’un modèle avec les structures connues. • Évolution: permet de révéler des relations évolutives non identifiable par l’alignement de séquence seulement.
Comparaison et Alignement de structure • Alignement ≠ Superposition • L’alignement c’est l’identification de résidus qui possèdent un même arrangement spatiale (3D). • La superposition nécessite au préalable l’alignement de résidus pour pouvoir superposer le reste de la structure…un genre de nœud de transformation géométrique. Chaîne A Chaîne B alignement superposition
Comparaison et Alignement de structure • Comment mesure-t-on l’erreur de superposition? • RMSD: root mean square deviation. Atome b Coordonnée (xb, yb, zb) Atome a Coordonnée (xa, ya, za) RMSD = S {(x – x)2+(y – y)2+(z – z )2 } ai bi ai bi ai bi i N Où N= nombre d’atomes comparés
Comparaison et Alignement de structure • 0.0-0.5 Å ⇒ Identique. • <1.5 Å ⇒ Très similaire. • < 5.0 Å ⇒ Modérément bon. • 5.0-7.0 Å ⇒ Relié structurellement. • > 7.0 Å ⇒ Relation incertaine. • > 12.0 Å ⇒ Aucunement relié.
CE exemple… Importance statistique p/r à un alignement avec une structure aléatoire. (conformation similaire: > 3.5) % de différence de longueur en a.a entre les structures alignées % de GAPs permis dans l’alignement des structures. Votre requête: utilisez le code pdb et précisez la chaîne avec << : >> (ex. 1A3G:A…fichier 1A3G chaîne A. Si il n’y pas de chaînes à spécifier, la notation est 1A3G:_ )
CE exemple… Faites vos sélections!
CE exemple… Lignes pointillées = Gap Lettrages pâles (très pâles…) = résidus non-alignés RMSD % d’identité # de résidus alignés Z-Score
CE exemple… Souris bouton droit Le fichiers sera en format .txt mais reconnu par Rasmol et Swiss PDBviewer… Nécessite l’installation du plug-in Chime/ protein explorer
CE exemple… RMSD = 0.5 Seq. Id. = 100.0 1A3G:A # résidus alignés = 274 Z-score = 7.5 1I1M:C
CE exemple… RMSD = 1.4 Seq. Id. = 27.3 1A3G:A # résidus alignés = 275 Z-score = 7.5 2DAB:B
CE exemple… RMSD = 2.0 Seq. Id. = 20.5 1A3G:A # résidus alignés = 260 Z-score = 7.2 1ET0:A
CE exemple… RMSD = 5.2 Seq. Id. = 5.2 1A3G:A # résidus alignés = 77 Z-score = 3.9 1I6H:E
CE exemple… RMSD = 6.0 Seq. Id. = 2.3 1A3G:A # résidus alignés = 83 Z-score = 3.9 1B76:B
CE exemple… RMSD = 8.6 Seq. Id. = 10.3 1A3G:A # résidus alignés = 107 Z-score = 3.7 1B76:B
VAST exemple… Faites vos sélections!
VAST exemple… À télécharger au préalable!