Download

1 / 43
450 likes | 737 Views
. . Utilizzare tessuti freschi o correttamente conservati. La QUALITA’ del materiale di partenza influenza la qualità e la resa del DNA isolato. . . Evitare di agitare fortemente o di pipettare. Molecole grandi di DNA sono facilmente danneggiate dalle forze frizionali. . .

E N D
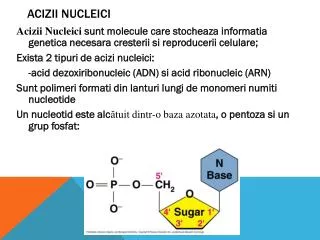
Utilizzare tessuti freschi o correttamente conservati La QUALITA’ del materiale di partenza influenza la qualità e la resa del DNA isolato Evitare di agitare fortemente o di pipettare Molecole grandi di DNA sono facilmente danneggiate dalle forze frizionali Lavorare in sterilità per prevenire contaminazioni da nucleasi Autoclavare soluzioni e strumenti Aggiungere alle soluzioni EDTA per chelare gli ioni Mg2+ necessari per l’attività delle DNasi Trattare materiali e soluzioni con DEPC per inibire le RNasi ISOLAMENTO degli ACIDI NUCLEICI AVVERTENZE:
PROCEDURA: Rottura dei tessuti (mortaio e pestello, omogeneizzatori…) Digestione enzimatica della parete cellulare e lisi della membrana plasmatica Trattamenti con RNasi e DNasi Deproteinizzazione mediante soluzioni acquose di fenolo/cloroformio Le proteine denaturate rimangono all’interfaccia fra la fase organica e quella acquosa Precipitazione con etanolo o isopropanolo Conservazione a -20°C/-80°C in un tampone contenente EDTA Il DNA è soggetto a idrolisi acida se conservato in acqua ...ISOLAMENTO degli ACIDI NUCLEICI
CONCENTRAZIONE: Misurazione dell’assorbimento a 260 nm [DNA]= 50 mg/ml [RNA]= 40 mg/ml A260 = 1 OD A260 = 1 OD PUREZZA: Rapporto A260/A280 A260/A280 = 1.7/1.9 (DNA) A260/A280 = 1.9/2.1 (RNA) Le misure spettrofotometriche non differenziano fra DNA ed RNA Il fenolo ha un massimo di assorbimento a 270-275 nm CARATTERIZZAZIONE SPETTROFOTOMETRICA
M 1 2 3 4 5 6 7 8 M Kb 1 2 3 4 5 97.0 48.5 28 S 26 S 18 S 25 S 23 S 18 S 16 S M: Markers 1: Sangue 2: Cellule HeLa 3: Cellule CHO 4: E. coli 5: B. subtilis 6: Coda di topo 7: Fegato 8: SS. cerevisiae 1: Topo 2: Uomo 3: Lievito 4: Batterio 5: Pianta CONTROLLO INTEGRITA’ e TAGLIA Elettroforesi su gel di agarosio
AAA AAA . . . . TTT . . . . . AAAA AAAA AAAA . TTTTT AAA AAA Oligo dT-cellulosa RNA cellulare totale . . TTT . L’mRNA con poli(A) si ibrida con l’oligo(dT) TTTTT . . . NaCl 100 mM . AAA AAAAA AAA AAA AAA TRIS 10 mM EDTA 1 mM L’rRNA ed il tRNA vengono eluiti Viene eluito l’mRNA ISOLAMENTO mRNA Solo gli mRNA possiedono code di poli(A) lunghe 30-150 residui. Le sequenze di oligo(dT) sono immobilizzate su supporti solidi, generalmente di cellulosa. L’RNA viene denaturato e quindi applicato alla colonna in una soluzione salina concentrata (NaCl 0.5 M). Si effettuano molti lavaggi con una soluzione salina meno concentrata (NaCl 100 mM) per rendere più selettivo il legame tra RNA e oligo(dT). Aggiungendo una soluzione di TRIDS/EDTA si recupera l’mRNA legato alla colonna.
Curva di fusione 1.3 1.2 1.1 1.0 Grado di denaturazione (%) 50 0 Assorbanza a 260 nm Tm 40 60 80 100 Temperatura (°C) ANALISI FISICA Effetto ipercromico: Aumento dell’A260nm durante il processo di denaturazione del DNA. Temperatura di melting (Tm): Temperatura in corrispondenza della quale il DNA è denaturato al 50%. Maggiore è G+C, maggiore è Tm.
Curva C0t Effetto ipocromico: Diminuzione dell’A260nm durante il processo di rinaturazione del DNA. Parametri che influenzano la rinaturazione: 1) C0 Concentrazione espressa in nucleotidi/unità di volume 2) Tempo La misura della velocità di rinaturazione può fornire utili informazioni circa la complessità del DNA 0 50 100 DNA a rinaturazione rapida Rinaturazione (%) DNA a rinaturazione lenta -2 -1 0 1 2 3 4 5 Log Cot CINETICA di RINATURAZIONE
Ha reso possibile il CLONAGGIO dei GENIpermettendo di ISOLARE AMPLIFICARE frammenti di DNA SEQUENZIARE TECNOLOGIA DEL DNA RICOMBINANTE Perché manipolare i geni? 1) Per facilitare lo studio dell’espressione genica e della regolazione fisiologica; 2) per identificare il prodotto di un gene e/o per ottenerne la sovraespressione; 3) per studiare la relazione fra struttura e funzione delle proteine; 4) per identificare componenti cellulari che interagiscono con particolari sequenze di acidi nucleici o con particolari domini proteici
PROCEDURA di CLONAGGIO ISOLAMENTO del gene INSERZIONE del gene in un VETTORE PLASMIDICO INTRODUZIONE del vettore plasmidico IN CELLULE VIVENTI per propagarlo Le fasi più delicate sono quelle del taglio e dell’unione di sequenze di DNA in modo preciso…. …..tutto ciò si ottiene con l’ausilio di ENZIMI
Entrambe le catene hanno la stessa sequenza se lette in direzione 5’3’ 5’-GAATTC-3’ 3’-CTTAAG-5’ Si distinguono in due classi: Tagliano il DNA in siti adiacenti alla sequenza riconosciuta Enzimi di CLASSE I Tagliano il DNA all’interno della sequenza riconosciuta Enzimi di CLASSE II ENDONUCLEASI di RESTRIZIONE Sono enzimi che tagliano entrambi i filamenti della doppia elica del DNA in corrispondenza di specifiche sequenze. Riconoscono SEQUENZE PALINDROMICHE di 4 o 6 nucleotidi
5’-GTTAAC-3’ 3’-CAATTG-5’ 5’-GTT AAC-3’ 3’-CAA TTG-5’ HpaI 5’-GGATCC-3’ 3’-CCTAGG-5’ 5’-G GATCC-3’ 3’-CCTAG G-5’ BamHI ...ENDONUCLEASI di RESTRIZIONE Possono lasciare estremità: TRONCHE(o blunt) se il taglio cade al centro della sequenza riconosciuta SPORGENTI(o 5’/3’ protruding) dette anche ADESIVE (o sticky) se il taglio avviene a posizioni sfalsate sui due filamenti
E’ il processo di determinazione delle posizioni dei siti di taglio delle endonucleasi di restrizione all’interno di un pezzo di DNA. IMPIEGHI COMUNI: Distinzione di molecole di DNA della stessa lunghezza, ma con sequenze diverse senza doverle sequenziare. Individuazione di mutazioni geniche responsabili di malattie genetiche. Identificazione di un frammento di interesse da un digerito in base al suo peso molecolare. MAPPATURA di RESTRIZIONE
DIMENSIONE dei FRAMMENTI (Kb) TRATTAMENTO INTERPRETAZIONE Nessuna digestione 9 9 2 + 7 Enzima A 2 7 A B A 3 6 Enzima B 3 + 6 B A 3 6 A B A 1 + 2 + 6 2 1 6 Enzima A+ B B 2 + 3 + 4 2 4 3 ...MAPPATURA di RESTRIZIONE
1 2 3 4 5 6 7 8 9 10 11 12 M 1 2 3 4 5 6 7 8 9 10 11 12 IDENTIFICAZIONE del FRAMMENTO di INTERESSE mediante Southern blotting M Markers 7 SstI/NcoI 1 pBlueStar 8 HinDIII 2 XbaI/SstI 9 NcoI/HinDIII 3 XbaI 10 NcoI 4 BamHI/XbaI 11 NdeI/NcoI 5 NcoI/XbaI 12 BamHI/NcoI 6 NcoI
CARATTERISTICHE DELLA REAZIONE: Presenza di ATP 10° C o.n. oppure temperatura ambiente, poche ore P P P P P P P P La bassa temperatura è consigliata in quanto, diminuendo l’energia cinetica delle molecole, riduce la possibilità che le estremità appaiate si separino prima di venir stabilizzate dalla ligazione. OH OH OH EcoRI BamHI OH 5’-G AATTC-3’ 3’-CTTAA G-5’ 5’-G GATCC-3’ 3’-CCTAG G-5’ OH OH OH OH DNA LIGASI Forma legami covalenti fra il gruppo fosfato 5’ di una estremità ed il gruppo ossidrilico 3’ della catena adiacente. L’enzima comunemente utilizzato è la T4 DNA Ligasiperché stabile e poco costosa.
DNA LIGASI BamHI OH OH 5’-G GATCC-3’ 3’-CCTAG G-5’ OH OH OH OH OH OH FOSFATASI ALCALINA P P P P P P P P P P P P P P P P OH OH OH OH OH OH OH OH Blunt ends ligation oppure trattamento con Transferasi terminale Sticky ends ligation HpaI OH 5’-GTT AAC-3’ 3’-CAA TTG-5’ OH AAA OH + dATP OH AAA Sticky ends ligation TTT TTT + dTTP
EcoRI BamHI 5’-G AATTC-3’ 3’-CTTAA G-5’ OH OH 5’-G GATCC-3’ 3’-CCTAG G-5’ OH OH NUCLEASI S1 OH OH P P P P P P P P P P P P P P P P OH OH OH OH OH blunt ends ligation oppure legame con un linker OH -GGAATTCC CCTTAAGG- EcoRI OH digestione + OH sticky ends ligation DNA LIGASI
Piccole molecole di DNA batterico extracromosomico, circolare. In natura conferiscono vantaggi selettivi ai ceppi che li contengono. Ingegnerizzati per l’uso di laboratorio. Per essere un buon vettore di clonaggio, un plasmide deve avere: Dimensioni contenute Un sito di origine della replicazione di tipo batterico controllato in maniera “rilassata” o “stringente” Due geni che conferiscono resistenza a due antibiotici Siti unici di restrizione PLASMIDI
METODO CHIMICO METODO ELETTRICO Le cellule sono rese COMPETENTI mediante: trattamento a freddo con ioni Ca2+ trattamento a freddo con glicerolo Il DNA viene incorporato mediante: SHOCK TERMICO IMPULSO di CORRENTE ELETTRICA PIASTRAMENTO SU TERRENO SELETTIVO Quelle che hanno incorporato il plasmide con l’inserto possono essere individuate grazie alla perdita di una funzione metabolica INATTIVAZIONE INSERZIONALE TRASFORMAZIONE delle CELLULE BATTTERICHE Solo le cellule che hanno incorporato il plasmide formeranno delle colonie.
Tampone di velluto Pistramento per replica Incubazione Crescono solo le cellule contenenti il plasmide privo dell’inserto Colonie sviluppatesi su terreno contenente ampicillina Piastra con terreno contenente tetraciclina Recupero delle colonie contenenti il plasmide ricombinante dalle piastre con ampicillina ANALISI dei PLASMIDI RICOMBINANTI PIASTRAMENTO PER REPLICA
pcs gene che codifica per la b-galattosidasi interrotto dal sito di clonaggio multiplo (pcs) lacZ L’enzima b-galattosidasi è in grado di metabolizzare il substrato incolore X-Gal formando un composto di colore blu. Un ceppo batterico lac-, trasformato con il plasmide ricombinante pUC19, è in grado di metabolizzare il substrato X-Gal solo se il plasmide è privo dell’inserto e forma colonie di colore blu. Colonie di batteri lac+che contengono plasmidi senza inserto Colonie di batteri lac-che contengono plasmidi ricombinanti Piastra con terreno contenente X-Gal …ANALISI dei PLASMIDI RICOMBINANTI SCREENING BLU/BIANCO
INTERVALLO (Lag phase) I batteri vengono diluiti nella coltura iniziale; la divisione procede lentamente in quanto le cellule si stanno adattando al terreno fresco. 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0 4 - 5 ore Densità cellulare (OD600) FASE LOGARITMICA I batteri crescono esponenzialmente. 10 - 11 ore 0 5 10 15 20 Tempo (h) FASE STAZIONARIA La densità cellulare rimane costante. La coltura può anche entrare in una fase di declino in cui le cellule si lisano ed il DNA si degrada parzialmente. Curva di crescita di E. coli
1. RISOSPENSIONE 3. NEUTRALIZZAZIONE KAc E. coli NaOH / SDS RNasi Centrifugazione 2. LISI Supernatante contenente il DNA plasmidico Precipitato contenente DNA genomico, proteine, detriti cellulari DNA cromosomico DNA plasmidico PURIFICAZIONE del DNA PLASMIDICO LISI ALCALINA
DNA cromosomico DNA plasmidico Bromuro d’etidio Bromuro d’etidio Svolgimento della doppia elica e diminuzione della densità idrodinamica Svolgimento della doppia elica compensato dall’introduzione di supereliche ...PURIFICAZIONE del DNA PLASMIDICO ULTRACENTRIFUGAZIONE in gradiente di densita’ di CsCl in presenza di EtBr Il superavvolgimento indotto nel DNA plasmidico dall’intercalazione del bromuro d’etidio fra le basi ne impedisce progressivamente il legame rendendo la densità idrodinamica del DNA plasmidico maggiore di quella del DNA cromosomico.
OTTENERE NUMEROSE COPIE IDENTICHE di un certo frammento di DNA CLONARE MA, OTTENERE DISCRETE QUANTITÀ del PRODOTTO PROTEICO codificato dal gene di interesse CLONARE IN UN VETTORE DI ESPRESSIONE VETTORI di ESPRESSIONE INSULINA ORMONE DELLA CRESCITA UMANO INTERFERONI sono solo pochi esempi di proteine commercializzate prodotte da batteri
SEQUENZA PROMOTRICE SEQUENZA SHINE-DALGARNO a monte di uno o più siti di inserzione del DNA estraneo AVVERTENZE: UTILIZZARE il cDNA CLONARE il cDNA nella CORRETTA CORNICE di LETTURA CONSIDERAZIONI: I batteri non sono in grado di eseguire modificazioni post-traduzionali ...VETTORI di ESPRESSIONE PROPRIETA’: Oltre alle normali caratteristiche di un vettore di clonaggio, un vettore per l’espressione di geni in cellule batteriche DEVE POSSEDERE:
IPTG IPTG E. coli RNA polimerasi T7 RNA polimerasi T7 RNA polimerasi T7 gene 1 gene target promotore lac lac o promotore T7 lac o DE3 repressore lac pET gene lac I repressore lac gene lac I CELLULA OSPITE genoma di E. coli ESPRESSIONE di GENI ETEROLOGHI in sistemi batterici Alcuni ceppi ospite sono stati opportunamente ingegnerizzati per l’espressione regolata dei geni. Per esempio, viene spesso usato un sistema che accoppia al sistema lac di E. coli una RNA polimerasi di batteriofago.
Citichinina STRATEGIA: Auxina Opina Sostituire i geni che causano il tumore con il gene di interesse, lasciando intatti i confini dx e sx necessari all’integrazione del DNA nel genoma della pianta. Confine sinistro Plasmide Ti Catabolismo dell’opina ori Geni vir Introdurre nel plasmidio: un gene marcatore selezionabile, sia vegetale che batterico; un’origine di replicazione che consenta al plasmidio di duplicarsi in E. coli ; un sito di clonaggio multiplo compreso fra i confini dx e sx. T-DNA PLASMIDI per la TRASFORMAZIONE GENETICA delle PIANTE Derivano da modificazioni del plasmide naturale Ti (Tumor inducing) del batterio del suolo A. tumefaciens che trasforma geneticamente le piante con un processo che rientra normalmente nel suo ciclo vitale. Confine destro
Confine destro Gene marcatore per E. coli e A. tumefaciens Gene marcatore selezionabile vegetale Gene bersaglio Gene marcatore selezionabile vegetale Confine destro Vettore cointegrato Confine sinistro E. coli ori Vettore binario Gene bersaglio Gene marcatore per E. coli e A. tumefaciens RICOMBINAZIONE Sequenza DNA omologa E. coli ori A. tumefaciens ori Confine sinistro Geni vir Plasmide Ti disarmato A. tumefaciens ori ...PLASMIDI per la TRASFORMAZIONE GENETICA delle PIANTE SISTEMA VETTORE BINARIO SISTEMA VETTORE COINTEGRATO Un plasmide Ti modificato contenuto in A. tumefaciens reca i geni vir, indispensabili all’integrazione del DNA compreso fra i confini dx e sx nel genoma della pianta.
Trasformazione mediata da A. tumefaciens Il nuovo plasmide entra nelle cellule dal bordo del ritaglio Dischetti di foglie poste su una sospensione di cellule di Agrobacterium Dischetti di foglie cresciute su un terreno nutriente selettivo; solo le cellule trasformate si moltiplicano Vengono riprodotte intere piante contenenti il gene introdotto Elettroporazione Alto voltaggio protoplasti Bombardamento con microproiettili ➺ ➺ ➺ ➺ ➺ ➺ ➺ Microproiettili di oro o tungsteno rivestiti di DNA plasmidico TRASFORMAZIONE GENETICA delle PIANTE
I plasmidi sono i vettori di elezione per clonare piccoli frammenti di DNA (5-10 Kb). INFATTI, Plasmidi di grandi dimensioni sono instabili e possono andare incontro a delezioni spontanee. La trasformazione non avviene efficentemente. QUINDI, Per clonare frammenti più grandi si impiegano come vettori del DNA dei batteriofagi (virus batterici)che consentono di iniettare grosse molecole di DNA nelle cellule batteriche per infezione. VETTORI FAGICI VETTORI COSMIDICI OLTRE AI PLASMIDI...
Un vettore comunemente utilizzato è il batteriofago l, lungo 49 Kb. Eliminazione del DNA non essenziale DNA virale Regione non essenziale (circa 20 Kb) DNA ricombinante DNA da clonare Assemblaggio in vitro delle particelle virali Teste e code del virus Il DNA virale entra nelle cellule, viene replicato e dirige la sintesi delle proteine virali. La lisi delle cellule rilascia le nuove particelle virali assemblate Infezione dei batteri Recupero delle particelle virali e isolamento del DNA clonato DNA VIRALE come VETTORE DI CLONAGGIO
COSMIDI Sono PLASMIDI, ma contengono, oltre alle caratteristiche essenziali di un plasmide, anche un SITO COS La presenza di questo elemento è determinante per consentire l’inserimento della molecola di DNA ricombinante nella testa del virus che, a sua volta, la inietta in una cellula batterica per infezione.
• Estremità coesive Sito cos Estremità coesive All’interno della cellula, le estremità si appaiano originando una molecola di DNA circolare Il DNA virale a doppia elica entra nelle cellule batteriche durante l’infezione come molecola lineare Cicli ripetuti di replicazione con il meccanismo del cerchio rotante Concatameri di copie di DNA virale 5’ Il DNA codifica per le proteine della testa e della coda del virus Il DNA viene inserito nelle teste del virus e tagliato a livello del sito cos Sono aggiunte le code Particella virale infettiva. I virus sono rilasciati per lisi delle cellule e possono infettare altre cellule batteriche. IMPACCHETTAMENTO del DNA VIRALE
Sito cos DNA da clonare Gene per la resistenza all’antibiotico Sito di restrizione ori Taglio del plasmide e del DNA da clonare con lo stesso enzima di restrizione Ligasi (i frammenti sono inseriti a caso fra due cosmidi) Impacchettamento in vitro solo se la distanza fra i due siti cos è compresa fra 37 e 52 Kb Infezione di E. coli con i fagi e selezione delle colonie resistenti all’antibiotico Batterio Il DNA ricircolarizza attraverso i siti cos e viene replicato all’interno del batterio come un plasmide VETTORI COSMIDICI
PROCEDURA: Isolamento del DNA genomico Restrizione parziale con un enzima che riconosca sequenze tetranucleotidiche Frammenti di 10-40 Kb Clonaggio in un vettore Bisogna produrre un numero discreto di cloni per far sì che ogni singolo gene sia presente almeno in un clone. Per il clonaggio di frammenti di centinaia di migliaia di basi si utilizzano come vettori i cromosomi artificiali di lievito. COSTRUZIONE BIBLIOTECHE GENOMICHE
Caratteristiche dei plasmidi: Origine di replicazione autonoma Geni marcatori di selezione Siti di clonaggio Centromero Sequenze telomeriche CROMOSOMI ARTIFICIALI di LIEVITO
AAA AAA AAAA AAAA AAAA AAA AAA PROCEDURA: Isolamento RNA messaggero Sintesi DNA complementare Trascrittasi inversa 7-mG AAAAA-3’ Clonaggio in un vettore AAA TTTT-5’ Questo tipo di biblioteche sono particolarmente utili per l’espressione dei geni tessuto-specifici. COSTRUZIONE BIBLIOTECHE di cDNA
Oligo (dT) TTTTT mRNA 7-mG AAAAA-3’ Trascrittasi inversa dATP, dCTP, dTTP, dGTP 7-mG AAAAA-3’ TTTT-5’ OH 7-mG AAAAA-3’ TTTT-5’ TTTT-5’ 7-mG 7-mG AAAAA-3’ AAAAA-3’ OH-3’ OH-3’ Frammento di Klenow dATP, dCTP, dTTP, dGTP 7-mG AAAAA-3’ TTTT-5’ TTTT-5’ Rnasi H Nucleasi S1 TTTT-5’ TTTT-5’ cDNA incompleto cDNA completo SINTESI cDNA
Placche di lisi Replica su filtri di nitrocellulosa (conservare le piastre originali) • • • • • • Denaturazione, neutralizzazione elegame del DNA al filtro Punti di riferimento per l’orientazione della replica al termine dell’esperimento • • • • Recupero delle placche positive dalla piastra originale • • • • • Sonda ibridizzata Autoradiogramma che mostra la posizione delle sonde ibridizzate Autoradiografia Ibridizzazione con sonda marcata e lavaggio della sonda non legata 32P 32P 32P 32P 32P 32P Sonda non legata SCREENING mediante IBRIDIZZAZIONE SU PLACCA
Una sonda è un filamento di DNA complementare a quello che si vuole isolare dalla biblioteca. STRATEGIE DI SINTESI: CASO 1: CASO 2: E’ nota la sequenza nucleotidica del gene di interesse E’ nota la sequenza amminoacidica della proteina codificata dal gene Si predice la sequenza nucleotidica che dovrebbe codificare per quella proteina Si sintetizzano chimicamente le sequenze nucleotidiche (degenerate) più appropriate o si amplifica un frammento del gene di interesse mediante PCR Nello screening di una libreria genica il cDNA può essere utilizzato come sonda per l’isolamento del corrispondente gene. SONDE OLIGONUCLEOTIDICHE
5’- -3’ DNA 3’- -5’ G C G C A A G 5’- -3’ DNA denaturazione 3’- -5’ C G C G T T C 3’-TAGCAG-5’ aggiunta di inneschi esanucleotidici 3’-GCATGC-5’ taglio di un’elica e rimozione di un nucleotide mediante la DNA polimerasi I 5’-TGCAGT-3’ ibridizzazazione 5’-GCATAC-3’ G G C A A G Pol I 5’- -3’ -5’ 3’- -3’ 5’- C G C G T T C 3’-TAGCAG-5’ 3’-TAGCAG-5’ riempimento del buco prodotto con un nucleotide radioattivo e rimozione del nucleotide successivo 32P-dCTP 5’-TGCAGT-3’ 5’-GCATAC-3’ 3’- -5’ G C C A A G -3’ 5’- dATP Frammento di Klenow, dNTP -5’ 3’- 32P-dCTP C G C G T T C dTTP dGTP il taglio si muove in direzione 5’-3’ dGTP Sintesi DNA -3’ 5’- G C G A A G 5’- -3’ 3’-TAGCAG-5’ 3’-TAGCAG-5’ 3’- -5’ DNA sonda marcato C G C G T T C 5’-TGCAGT-3’ 5’-GCATAC-3’ 32P-dCTP 3’- -5’ denaturazione G C G C A G 5’- -3’ 3’- -5’ C G C G T T C MARCATURA delle SONDE OLIGONUCLEOTIDICHE RANDOM PRIMING Metodo dell’innesco casuale NICK TRANSLATION Spostamento dell’incisione 3’- -5’