Download

1 / 16
170 likes | 573 Views
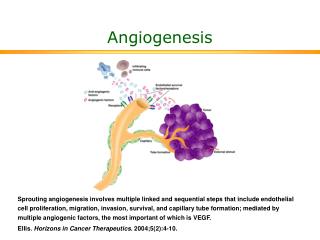
VEGF 及其信号转导途径. 目录. 1 . VEGF 的功能 2 . VEGF 的受体 3. 刺激 VEGF 表达和分泌的因素 4 . VEGF 诱发的细胞内信号转导 5 . VEGF 通过的各信号转导通路间的相互作用. 1 . VEGF 的功能. 血管基底膜的降解、血管内皮细胞 (EC) 的增生、移行以及新基底膜的沉积等一系列反应 , 最终导致新生血管形成 目录. 2 . VEGF 的受体.

E N D
目录 • 1. VEGF的功能 • 2. VEGF的受体 • 3. 刺激VEGF表达和分泌的因素 • 4.VEGF诱发的细胞内信号转导 • 5.VEGF通过的各信号转导通路间的相互作用
1. VEGF的功能 • 血管基底膜的降解、血管内皮细胞(EC)的增生、移行以及新基底膜的沉积等一系列反应,最终导致新生血管形成 • 目录
2. VEGF的受体 • VEGF受体的3个亚型中, VEGFR-1/Flt-1和VEGFR-2/KDR/Flk-1可表达于EC。VEGFR3是特异表达在淋巴内皮细胞中。VEGF的生物学效应主要是通过VEGFR-2实现。Flt-1虽能结合VEGF,但不参与血管形成相关信号的转导,属“诱骗受体”,竞争性抑制KDR的生物学效应。Neuropilin-1(NP-1)是KDR的辅助受体,使KDR与VEGF165的结合增加。 • 目录
3.刺激VEGF表达和分泌的因素 • 能够合成和分泌VEGF的细胞有EC、MP、Müller细胞、成纤维细胞和RPE细胞等。诱导合成和分泌VEGF的因素包括炎症、缺氧、高度糖基化终产物(AGEs)和细胞外基质异常等。
3.刺激VEGF表达和分泌的因素 • 3.1 炎性细胞分泌的TGF、bFGF、PDGF-β、胰岛素样生长因子和白细胞介素-1(IL-1)等细胞因子对VEGF的表达有正调节作用。MP在血管新生过程中起着关键性作用。 • 3.2 在缺氧环境中,VEGF启动子被激活的同时,与磷酸化的缺氧诱导因子-1α(HIF-1α)结合,增强了VEGF基因的转录活性。应激激活蛋白激酶则可保持VEGF mRNA的稳定。此外,VEGF的DNA序列上有氧敏感基因——促红素基因,直接调节VEGF的表达。缺氧下调某些血管生成抑制剂的表达,也导致VEGF相对增加。 • 3.3 AGEs可诱导EC胞内蛋白激酶Cβ(PKCβ)的转移及细胞外信号调节激酶1/2(ERK1/2)和核因子-κB(NF-κB)的活化,故推测AGEs诱导的VEGF表达可能通过PKC、丝裂原激活的蛋白激酶(MAPK)和NF-κB通路实现 • 3.4 除对缺氧和炎症介质敏感外,将能够合成和分泌VEGF的细胞暴露于异常的细胞外基质蛋白中(如血小板反应素1),也能诱导VEGF的表达。诱导产生的VEGF再作用于该细胞,可引起该细胞DNA合成增加和VEGF的表达(VEGF的自分泌环),形成VEGF分泌的正反馈。 • 目录
4.VEGF诱发的细胞内信号转导 • 4.1 丝裂原活化的蛋白激酶(MAPK)途径 • 4.2 PI-3K-AKT/PKB途径 • 4.3 Ca2+-磷脂依赖性激酶途径 • 4.4 NO途径 • 4.5 示意图
VEGFR2介导的信号转导的主要过程是:VEGF结合到VEGFR2胞外区的Ig结构域上,引起VEGFR2胞内激酶区特定酪氨酸残基的交叉磷酸化而活化,VEGFR-2已有6个自身磷酸化位点被证实,Tyr-951、Tyr-996、Tyr-1054、Tyr-1059、Tyr-1175、Tyr1214。活化的受体进而通过一系列生化级联反应启动特定基因的表达,完成VEGF相应的促增殖作用。
4.1 丝裂原活化的蛋白激酶(MAPK)途径 • VEGF结合到VEGFR2胞外区的Ig结构域上,引起KDR活化,活化的KDR 通过自身磷酸化位点Tyr-951与接头蛋白Grb2,Grb2通过SH2结构域结合鸟苷酸交换蛋白(SOS),使之接近并活化Ras,然后进一步激活MAPK级联反应:Raf-1→MEK1→ERK1/2。然而也有人认为,VEGF是以一种不依赖Ras而由蛋白激酶C(PKC)介导的方式激活MAPK级联反应的。活化的ERK1/2可通过诱导P90核糖体S6激酶(P90RSK)磷酸化来实现其生物学效应。ERK1/2的下游转录因子可能是ets-1。ets-1识别GGA(A/T) DNA序列,控制c-ets的转录。除ERK外,VEGF诱导活化的另一个MAPK是C-Jun末端蛋白激酶(JNK)。VEGF诱导的有丝分裂需要ERK和JNK通路的相互作用。 • VEGF可诱导P38-MAPK活化,P38-MAPK继而活化MAPKAP激酶-2/3,并使丝状肌动蛋白(F-Actin)聚合调节分子和热休克蛋白27(HSP27)发生磷酸化[33],引起肌动蛋白细胞骨架的重组,产生EC移行。
4.2 PI-3K-AKT/PKB途径 • VEGF可通过PI-3K/AKT通路诱导EC增生和移行。VEGF通过整合素相关激酶(ILK)诱导AKT/PKB的磷酸化。AKT活化后,AKT/PKB蛋白的降解受到抑制,增强其丝/苏氨酸激酶的稳定性。AKT的下游可能为BAD、天冬氨酸特异性半胱氨酸蛋白酶(caspase)或forkhead转录因子。磷酸化的AKT可抑制BAD和caspase-9的活化,发挥抗EC凋亡的作用。
4.3 Ca2+-磷脂依赖性激酶途径 • VEGF活化磷脂酶Cγ(PLCγ),PLCγ水解膜组分磷脂酰肌醇-4,5-二磷酸(PIP2)产生DAG。同时Ca2+使PKCα、PKCζ结合并聚集至质膜,在二脂酰甘油(DAG)的作用下活化。PKC的下游可能是NF-κB。PKC也可作为内皮一氧化氮合酶(eNOS)和Raf-ERK1/2 MAPK的上游激活物,参与不同的信号转导通路。
4 .4 NO途径 VEGF刺激EC会引起NO的释放, NO的释放可引起PKCδ活性的下降。关于NO如何降低PKCδ的活性有两种推测:(1)很多PKC亚型的活性可通过丝氨酸-苏氨酸磷酸化来调节,NO可活化cGMP-依赖性蛋白激酶(PKG),故NO可能通过它降低PKCδ的活性;(2)PKCδ催化域中有一个酪氨酸可被磷酸化,可通过某种酪氨酸激酶(PTK)改变其活性。PKCδ的活性降低抑制周期蛋白依赖激酶2改变细胞周期进程或影响Ras下游的信号转导引起EC增生和移行。
5.VEGF通过的各信号转导通路间的相互作用 CEC的增生需要MAPK/ERK与PI-3K/AKT双信号通路,两信号通路共同的上游信号分子是Ras。PI-3K除作用于AKT外,还位于ERK1/2上游,控制ERK通路。VEGF与KDR结合后,PLD2与Ras发生聚集。使用PLD抑制剂可抑制ERK的磷酸化,同时也抑制了VEGF诱导的EC增生。 目录