Download

1 / 31
310 likes | 494 Views
CASP 5. Fifth Meeting on the Critical Assessment of Techniques for Protein Structure Prediction Robert Langlois. Purpose. Establish in structure prediction from sequence Capabilities Limitations Accomplished by analysis of a large number of blind predictions. Prediction Categories.

E N D
CASP 5 Fifth Meeting on the Critical Assessment of Techniques for Protein Structure Prediction Robert Langlois
Purpose • Establish in structure prediction from sequence • Capabilities • Limitations • Accomplished by analysis of a large number of blind predictions
Prediction Categories • CASP 5 • Comparative Modeling • Fold Recognition • New Fold methods • CAFASP 3 • Automated Servers
Category Target Overlap Comparative Modeling Fold Recognition Ab Initio
Comparative Modeling • Exploit evolutionary relationships to produce 3D structures • Has not changed in a few decades • General Protocol • Start by identifying the template • Align sequences of target and template • Model conserved then diverged regions • Assign side chain conformations, refine model
Scoring • GDT-TS • Global distance test • Number of Cα in prediction not deviating more than di from Cα in the target • Under the condition of optimal super-position • RMSD of Cα • Percent of Correctly Aligned Residues
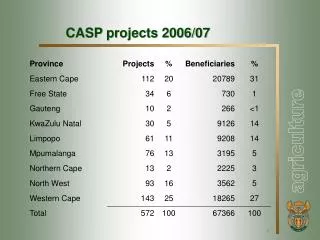
Comparative Modeling Results • Over 39 proteins, consisting of 51 domains • Able to identify templates with 6% identity • Improvement of CASP4, <17% identity • Top 5 groups – Initial predictions • Murzin: Knowledge-based personal approach • Bujnicki-Janusz: Automatic servers • VENCLOVAS: Multiple sequence alignment • Ginalski: Automatic servers • GeneSilico
Group Results cont. 448: Murzin 425: VENCLOVAS 020: Bujnicki 453: Ginalski 517: GeneSilico
Overall Results • Servers better than most human predictors • 3D Shotgun meta-predictor • Baker’s ROBETTA server • However, no group was able to optimize • I.e. create many good models but cannot pick the best • No model comes significantly closer to target than the template
CM Conclusion • Successes • Matching template to target • Performance of different methods has leveled • Failures • Cannot produce model, closer than template • Cannot model features not inherited • Future: select the best of several models
Fold Recognition • Common approaches: taxonometric (SVM, NN.), threading, homology modeling • Combines fold recognition, comparative modeling, and de novo approaches
Fold Recognition Methods • Template based combines • Correct template, comparative modeling and fold recognition servers • Refinement, available programs, and manual inspection • Ginalski • Fragment Assembly: Ab initio • Rosetta: identify small fragments from a library of existing structures • TOUCHSTONE: conserved contacts & threading
Scoring • Livermore: GDT_TS, SOV_O, and LGA_Q • 3 structural superposition, 2 sequence depend • Incorporate scores from: • Dali: http://www.ebi.ac.uk/dali/ • CE: http://cl.sdsc.edu/ce.html • Mammoth: http://icb.mssm.edu/services/… • Dali & CE compare intra molecular Cα geometries • Mammoth: structural alignments independent of contact maps, depend on unit vector RMSD distances
Fold Recognition Results • Top 3: Outperform the Rest • Baker – comparative modeling and ab initio • Ginalski – combine servers and manual inspection • Rychlewski – meta-server 3D Jury
Conclusions • While overall scores are higher than CASP4 • Does it reflect: better predictions or larger database? • Automatic servers nearly reach manual predictions
New Fold Techniques • Ab Initio Folding Engine • Metropolis Rule • Potentials • Successful Groups use: • Fragment Methods • Contact Maps
Scoring • RMSD, LCS, SOV, GDT_TS, Visual • GDT_TS agreed best with visual inspection • LCS – CASP3 holdover • Sequence dependent structural alignment
Discussion • Fragment methods: better at choosing fragments than assembling them. • Automatic Servers similar performance • PROTINFO-AB, I-site/Bystroff, BAKER-ROBETTA • 20 out of 25 groups use PSI-PRED
Conclusions • Coordinate Predictions • Impressive accuracy • Shows great progress in understanding folds • Convergence of fold recognition • Fragment: template library construction • Secondary Structure • Have reached limits
Conclusions cont. • Residue-Residue Contacts • Very limited improvement • Fragment templates, better accuracy • Not accurate enough to build a model from scratch
References Aloy, P., A. Stark, et al. (2003). "Predictions Without Templates: New Folds, Secondary Structure, and Contacts in CASP5." PROTEINS: Structure, Function, and Genetics 53: 436-456. Kinch, L. N., J. O. Wrabl, et al. (2003). "CASP5 Assessment of Fold Recognition Target Predictions." PROTEINS: Structure, Function, and Genetics 53: 395-409. Tramontano, A. and V. Morea (2003). "Assessment of Homolgy-Based Predictions in CASP5." PROTEINS: Structure, Function, and Genetics 53: 352-368.













![CompTIA CASP RC0-C02 CASP Practice Questions [2019 Updated]](https://cdn4.slideserve.com/8134489/comptia-advanced-security-practitioner-rc0-c02-dt.jpg)




