Download

1 / 55
570 likes | 793 Views
Inherited Diseases of the Glomerular Basement Membrane. Scope. Introduction Inherited diseases of glomerular basement membrane Description – clinical-pathological features, treatment Summary Key points. Introduction. The glomerular basement membrane (GBM)

E N D
Scope • Introduction • Inherited diseases of glomerular basement membrane • Description – clinical-pathological features, treatment • Summary • Key points
Introduction • The glomerular basement membrane (GBM) • Unique type of basement membrane • Great thickness (300–350 nm) and • Position between two cell layers, podocytes and endothelial cells • Specific role in maintenance of the glomerular filtration barrier Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Introduction (contd) • Major GBM components • Type IV collagen • Laminin • Nidogen (entactin) and • Heparan sulfate proteoglycans (HSPGs) • Mutations in • type IV collagen or laminin genes have been shown to be associated with hereditary glomerular diseases Eur J Biochem 1989;180: 487–502.
Hereditary Diseases of Type IV Collagen • Alport syndrome • Characterized by the familial occurrence of progressive hematuric nephritis and hearing loss • Depending on the population studied, Alport syndrome affects 0.3–2.3% of all patients who develop end-stage renal disease (ESRD) in Europe, India or the US Br Med J 1927; 1: 504–6
Hereditary Diseases of Type IV Collagen • Alport syndrome (contd) • Hematuria—macroscopic/microscopic— is the principal and constant feature • Early-onset symptom, usually detected in childhood • Episodes of macroscopic hematuria precipitated by exercise/upper respiratory tract infection are observed in about 60% of patients < 15 years Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Hereditary Diseases of Type IV Collagen • Alport syndrome(contd) • Proteinuria increasing with age and progressive renal failure are observed, depending on the sex of the patient and the mode of transmission of the disorder • Hypertension does not usually develop before the onset of chronic renal insufficiency • Bilateral sensorineural hearing loss affecting high and middle frequencies is never congenital in Alport syndrome, but it can be detected during the first decade of life Am J Med 1981 70: 493–505, Am J Kidney Dis 1993; 22: 627–40, Acta Paediatr 1996; 85: 1300–6
Hereditary Diseases of Type IV Collagen • Alport syndrome (contd) • Anterior lenticonus is a conical protrusion of the anterior aspect of the lens that develops progressively • Retinal changes are characterized by the progressive appearance of asymptomatic perimacular yellowish flecks • Both types of ocular lesion are specific to Alport syndrome, and are observed in about 1/3 of patients • Detecting such lesions can, therefore, be useful for diagnosis of the disorder • Nonspecific lesions of the cornea have also been reported Clin Nephrol 1980;13: 163–7
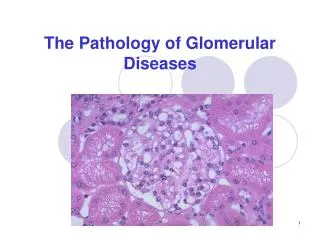
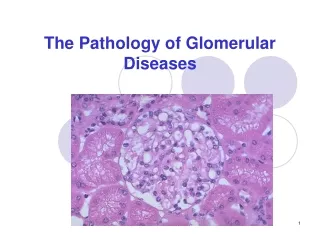
Hereditary Diseases of Type IV Collagen Electron microscopy images of renal tissue from patients with Alport syndrome. (A) Thickening and splitting of the GBM in an 11-year-old patient. The inner and outer contours of the GBM are ‘festooned’. Magnification ×30,000. (B) In tissue from a 12-year-old patient observed under low magnification, the irregular thickness of the GBM is evident. Magnification ×5,400. (C) Diffuse thinning of the GBM in a 3-year-old male patient. Magnification ×6,200. All sections stained with uranyl acetate and lead citrate. Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Immunohistological analysis of the renal distribution of type IV collagen chains. The analysis was carried out in (A–D) control, X‑linked (E,F) male and (G,H) female Alport syndrome patients, and (I–L) patients with autosomal recessive Alport syndrome, using antibodies to α1(IV) (A,E,I), α3(IV) (B,F,J), or α5(IV) (C,G,K,L) chains. Double labeling was made with anti-α2(IV) in red, and anti-α5(IV) in green (D,H). In control kidney, the α1(IV) chain is present in the mesangial matrix, Bowman’s capsule, and the extraglomerular basement membranes (A). The α3(IV) and α5(IV) chains are distributed within the GBM (B and C, respectively). The Bowman’s capsule is strongly α5(IV)‑positive (C). In X-linked Alport syndrome, no α3(IV) expression was detected in a male patient (id for α4–α5) (F), whereas the distribution is segmental in a female patient (G). In autosomal recessive Alport syndrome, no α3(IV)–α5(IV) labeling is detected in the GBM (J) whereas α5(IV) is expressed in Bowman’s capsule (K) and the basement membranes of the collecting ducts (L). In both types of Alport syndrome, α1(IV) is diffusely expressed in the GBM (E,I). Abbreviation: GBM, glomerular basement membrane Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Hereditary Diseases of Type IV Collagen • X-linked Dominant Alport Syndrome • In about 85% of affected families of European origin, Alport syndrome is transmitted as an X-linked dominant trait • This mode of transmission is characterized by • Greater disease severity in males > females and • Absence of father-to-son transmission Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Hereditary Diseases of Type IV Collagen • X-linked Dominant Alport Syndrome (contd) • Two forms on the basis of the rate of progression • 'juvenile' type • Highly stereotypical course within each affected family and by the occurrence of ESRD in men at about 20 years of age; • 'nonprogressive' or 'adult' type • ESRD develops at approximately 40 years of age and the disease course is much more variable Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Hereditary Diseases of Type IV Collagen • X-linked Dominant Alport Syndrome (contd) • Diffuse leiomyomatosis • Associated with Alport syndrome in 2–5% of families with the juvenile form of the disease • Affects the esophagus, the tracheobronchial tree and the female genital tract • Completely penetrant and fully expressed, even in female patients with mild renal disease Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Hereditary Diseases of Type IV Collagen • X-linked Dominant Alport Syndrome (contd) • Immunohistological analysis of the distribution of the different chains of type IV collagen in the basement membranes where COL4A5 is normally expressed is of the utmost importance in the diagnosis of Alport syndrome and the recognition of X-linked transmission • In most patients, the α5(IV) chain defect (absence or abnormal structure as a result of mutation) impairs protomer assembly and the formation of normal collagen IV networks Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Hereditary Diseases of Type IV Collagen • X-linked Dominant Alport Syndrome (contd) • Within the kidney, abnormal distribution of the α5(IV) chain is observed in approximately two-thirds of patients with X-linked Alport syndrome: • The α5(IV) antigen is absent from the glomerular, capsular and distal tubular basement membranes in male patients, and has a discontinuous distribution in related female patients Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Immunohistological analysis of the distribution of the α5(IV) collagen chain. The analysis was carried out in the skin of (A) controls, (B) male and (C) female patients with X-linked Alport syndrome, and (D) patients with autosomal recessive Alport syndrome. The α5(IV) chain is (A) present in the epidermal basement membrane (arrow) of the skin of controls, but is (B) absent from the skin basement membrane (arrow) of the male patient with X-linked Alport syndrome. (C) Segmental labeling (single arrow) is seen in the X-linked female patient. Nonspecific labeling of the keratin layer is indicated by a double arrow. (D) Normal staining patterns of the epidermal basement membrane (single arrow) are observed in autosomal recessive Alport syndrome. Nonspecific labeling of the keratin layer is indicated by a double arrow. Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Hereditary Diseases of Type IV Collagen • Autosomal Recessive Alport Syndrome • About 10–15% of families affected by the disease • Clinical and morphological features are identical in the autosomal recessive and X-linked forms • Usually severe • Nephritis progresses to early-onset ESRD • Hearing impairment affects the majority of patients, and • ocular lesions may or may not be present Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Hereditary Diseases of Type IV Collagen • Autosomal Recessive Alport Syndrome (contd) • Indicated by one/more of the following: • Severe disease in young females • Consanguinity in the family • Absence of severe renal disease in the parents of a patient • Microscopic hematuria in the father of an affected male; and • Immunohistochemical findings Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Hereditary Diseases of Type IV Collagen • Autosomal Dominant Alport Syndrome (contd) • Male-to-male transmission and similar disease severity in men and women, has been observed in a few families • The clinical phenotype is variable and milder than that of the X-linked dominant form • Progression to ESRD and hearing defect are not always seen and usually occur after 50 years of age • No ocular involvement has been reported Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Hereditary Diseases of Type IV Collagen • Benign Familial Hematuria (BFH) With Thinning of the GBM • The exact prevalence of is not known (estimates range from 1% to 10% of the population), but it is the most common cause of persistent hematuria • Inherited in an autosomal dominant manner • Characterized by familial occurrence of persistent or recurrent hematuria that is often detected in childhood Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Hereditary Diseases of Type IV Collagen • BFH (contd) • No significant proteinuria • No progression to renal failure and • No extrarenal symptoms are observed, and • The prognosis is excellent • GBM is uniformly thin • 'thin basement membrane nephropathy' (TBMN) is currently used as an alternative term for BFH Arch Intern Med 1973;131: 257–62
Hereditary Diseases of Type IV Collagen • BFH (contd) • GBM is uniformly thin • 'thin basement membrane nephropathy' (TBMN) is currently used as an alternative term for BFH • Use of a descriptive term (TBMN) for this disorder can cause confusion, • Because of the nonspecificity of thinning of the GBM, which can also be evident in patients with Alport syndrome, including adults Arch Intern Med 1973;131: 257–62
Hereditary Diseases of Type IV Collagen • BFH (contd) • Comprehensive familial investigations and regular follow-up assessments are of the utmost importance if BFH/TBMN is to be correctly distinguished from progressive nephritis • The onset of proteinuria or extrarenal symptoms should lead to reconsideration of the benign prognosis J Am Soc Nephrol 1998; 9: 1736–1750
Hereditary Diseases of Type IV Collagen • Treatment • Progression to ESRD is ineluctable in males with X-linked Alport syndrome and in all patients with the autosomal recessive form of the disease • Renal transplantation is generally a satisfactory treatment, but about 2.5% of patients develop anti-GBM glomerulonephritis leading to rapid graft loss Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Hereditary Diseases of Type IV Collagen • Treatment (contd) • Temporary or prolonged alleviation of proteinuria has been achieved by blockade of the renin–angiotensin system, but no data are available on the long-term evolution of renal function in response to this treatment • Controversy over the effects of ciclosporin in these patients Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Laminin β2 Disease • Pierson Syndrome • In 1963, Pierson et al. reported • The curious association in siblings of eye abnormalities with microcoria and congenital nephrotic syndrome progressing rapidly to ESRD • This association was also observed in a few neonates Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Laminin β2 Disease • Pierson Syndrome (contd) • Glomerular lesions were classified as mesangial sclerosis, with diffuse alteration of the GBM • Hypotonia and psychomotor retardation developed in the few patients who survived for several months after birth Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Laminin β2 Disease Laminin α5β2γ1. (A) Laminins are heterotrimeric molecules consisting of one α, one β, and one γ chain, with a cruciform organization. The major laminin of the glomerular basement membrane is α5β2γ1 (B) In the kidney, the laminin β2 chain is expressed at high levels in the glomerular basement membrane and in the basement membranes of arterial smooth muscle cells Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Laminin β2 Disease • Pierson Syndrome (contd) • This defect involves the LAMB2 gene encoding the β2 chain of laminin, which is expressed at high levels in the GBM, synaptic basal laminae and basement membranes of the eye • Recessive missense mutations have also been detected in two patients in a consanguineous family with isolated congenital nephrotic syndrome, a finding that expands the clinical spectrum of LAMB2-associated disorders Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Laminin β2 Disease • Pierson Syndrome (contd) • Mice lacking laminin β2 develop massive proteinuria, and their retinal and neuromuscular differentiation is abnormal • Interestingly, the onset of proteinuria precedes podocyte abnormalities, showing • GBM acts as a protein barrier and that the glomerular slit diaphragm alone is not sufficient to prevent the passage of albumin Nat Genet 1995; 10: 400–6; J Clin Invest 2006;16: 2272–9.
Hereditary Renal Diseases With Type III Collagen Deposits • Nail–patella Syndrome • Also k/as hereditary osteoonychodysplasia • Rare autosomal dominant disorder (affecting 1 in 50,000 individuals) • Characterized by an association between nail hypoplasia or dysplasia and bone abnormalities that primarily affect the knees, elbows and pelvis • Normal-tension glaucoma and sensorineural hearing impairment have been recognized as additional features J Med Genet 2005;40: 153–62
Hereditary Renal Diseases With Type III Collagen Deposits • Nail–patella Syndrome (contd) • Prognosis depends on the presence and severity of renal involvement, which is observed in 30–40% of patients • Renal involvement usually manifests as proteinuria, sometimes with hematuria • Progression to kidney failure occurs in about 30% of patients with renal symptoms, usually many years after the discovery of proteinuria, but in some cases during childhood Am J Pathol 2003;163: 145–155
Hereditary Renal Diseases With Type III Collagen Deposits • Nail–patella Syndrome (contd) • Light microscopy of renal tissue from people with NPS reveals no specific changes • The hallmark of the disease, observed by electron microscopy, is the presence of clusters of fibrillar type III collagen irregularly distributed within thick GBM segments and the mesangial matrix Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Hereditary Renal Diseases With Type III Collagen Deposits • Nail–patella Syndrome (contd) • Staining with phosphotungstic acid is often needed to reveal the collagen bundles; when standard staining techniques are used, the GBM has a mottled appearance • The extent and distribution of GBM lesions vary widely from case to case • There is no correlation between these microscopical features and patient age or the presence or severity of renal symptoms Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Hereditary Renal Diseases With Type III Collagen Deposits • Collagen Type III Glomerulopathy • Accumulation of type III collagen in the glomerular extracellular matrix has been detected in some proteinuric patients—mostly Japanese— in the absence of any other symptoms of NPS • In contrast to NPS glomerulopathy, diffuse glomerular changes (i.e. marked expansion of the mesangial matrix and thickening of the capillary walls) can be observed under the light microscope Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Hereditary Renal Diseases With Type III Collagen Deposits • Collagen Type III Glomerulopathy (contd) • At the ultrastructural level, the mesangial matrix and the subendothelial aspect of the GBM are enlarged and have a mottled appearance due to the presence of fibrillar collagen (which can be visualized after staining with phosphotungstic acid) • Unlike the lesions observed in NPS, the lamina densa is usually preserved Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Hereditary Renal Diseases With Type III Collagen Deposits • Collagen Type III Glomerulopathy (contd) • The clinical features of collagen type III glomerulopathy are highly variable • Two forms of the disease can be distinguished on the basis of age at onset of symptoms • In Japanese and a few Caucasian patients, the disease is usually sporadic and first symptoms—persistent proteinuria, with or without hypertension—are detected in adulthood Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Hereditary Renal Diseases With Type III Collagen Deposits • Collagen Type III Glomerulopathy (contd) • The severity of proteinuria and serum creatinine concentration increase slowly, and renal dysfunction is a late event • Glomeruli are strikingly enlarged as a result of massive deposits of type III collagen • An elevated serum level of the type III procollagen peptide seems to be a good marker for the disease, indicating increased synthesis of type III collagen, but the fundamental underlying defect remains unknown Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Hereditary Renal Diseases With Type III Collagen Deposits • Collagen Type III Glomerulopathy (contd) • Onset of first symptoms of collagen type III glomerulopathy in early childhood indicates autosomal recessive transmission of the disease • Under light microscopy, subendothelial enlargement of the capillary walls is moderate and might mimic diffuse thrombotic microangiopathy • This variant of the disease is severe Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Hereditary Renal Diseases With Type III Collagen Deposits • Collagen Type III Glomerulopathy (contd) • Collagen type III glomerulopathy is characterized by progressive worsening of proteinuria; nephrotic syndrome eventually develops, and early occurrence of hypertension and progression to renal failure are observed in most children • Anemia of the hemolytic type and unexplained respiratory symptoms have been reported, as has abrupt progression to ESRD resulting from superimposed hemolytic– uremic syndrome Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Hereditary Renal Diseases With Type III Collagen Deposits • Collagen Type III Glomerulopathy (contd) • Interestingly, collagen type III glomerulopathy has been reported in one patient who presented with an inherited factor H deficiency • Collagen type III glomerulopathy associated with factor H deficiency was also observed in our laboratory (unpublished data), in a young patient with consanguineous parents Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Hereditary Renal Diseases With Type III Collagen Deposits • Collagen Type III Glomerulopathy (contd) • Such findings indicate that factors that contribute to familial hemolytic– uremic syndromes might be involved in collagen type III glomerulopathy • These factors should be systematically evaluated in patients with the latter disorder Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Fibronectin Glomerulopathy • Fibronectin Glomerulopathy • Several familial cases of atypical lobular glomerulopathies, transmitted as autosomal dominant traits and • Characterized by the presence of massive parietal and mesangial fibrillar deposits of fibronectin, have been reported Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Fibronectin Glomerulopathy • Fibronectin Glomerulopathy (contd) • Patients have proteinuria, hematuria and hypertension, and progress slowly to ESRD • The fibronectin deposited in glomeruli is derived primarily from plasma, and recurrence in grafted kidneys has been observed Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Fibronectin Glomerulopathy • Fibronectin Glomerulopathy (contd) • Diagnosis can easily be established by immunohistochemical analysis of renal biopsy material, but • The pathophysiological role of fibronectin and the genetic cause of the disease are unknown Nat Clin Pract Nephrol CME. 2008;4(1):24-37.
Summary • Alport syndrome was the first characterized, and is the most frequent hereditary hematuric disorder of the GBM that progresses to ESRD • Analysis of the distribution of the collagen α5(IV) chain in skin is an important step in the diagnostic approach to the X-linked form of the disease
Summary (contd) • As technologies are improving rapidly, diagnosis based on DNA sequencing will become a more practical option in the near future • Therapeutic progress is expected to result from extension of the promising results obtained in animal models to human trials
Summary (contd) • Distinguishing between Alport syndrome and BFH/TBMN is sometimes difficult in young patients, in sporadic cases and in small families from which little useful information can be derived • Thinning of the GBM is not a marker of a specific disease entity, and does not guarantee a benign disease course
Summary (contd) • Long-term follow-up and repeat investigations might be needed before a definitive diagnosis can be made • The wide spectrum of phenotypes observed in patients that carry a heterozygous COL4A3 or COL4A4 mutation heralds a need for caution during risk assessment and genetic counseling