Download

1 / 33
340 likes | 591 Views
Glomerular diseases. 1. Normal anatomy 2. Pathogenesis 21 Hypersensitivity reactions 22 Pathogenetic mechanisms of glomerular diseases 23 Mechanisms of vascular injury. 1 Normal anatomy

E N D
Glomerular diseases • 1. Normal anatomy • 2. Pathogenesis 21 Hypersensitivity reactions 22 Pathogenetic mechanisms of glomerular diseases23 Mechanisms of vascular injury
1 Normal anatomy Fig. 1 Anatomy of the glomerulus and thejuxtaglomerular apparatus 1
Fig. 2 A podocyte surrounding a glomerular capillary All three layers (endothelium, glomerular basement membrane, slit pores between podocytes) are negatively charged Mesangium is contractable F 2
2 Pathogenesis • 21 Hypersensitivity reactions: • Mast cells covered with IgE antibodies bind parasite antigens inflammatory response, attraction of eosinophils killing worms
Type I: Immediate hypersensitivity (anaphylaxis) • 1st exposure to antigen production of specific AB their binding • to mast cells (sensitization). Next exposure allergen • degranulates mast cells release of histamine (immediate • response) vascular permeability, airways, hives, • conjunctivitis, rhinitis. Later: leukotriens, prostaglandins, PAF, • proteases (late phase) localizedanaphylaxis = atopy (asthma, • hay fever, eczema, hives) • systemic anaphylaxis – circulatory shock, • dyspnea, laryngospasm • Ts activity
Type II: Antibody-dependent cellular cytotoxicity (ADCC) • Cell-mediated cytotoxicity that requires prior binding of antibodies to • target cells • K(iller) cells: Lymphocyte-like cells (not B or T) that kill a variety of tumor cells and virus-infected cells but only after previous immunization • Errant or uncontrolled plasma cells produce antibodies against selfantigens • Drugs combine with body antigens (e.g., on erythrocytes) • anchor and activate K-cells ADCC • AB attach to the • surface of cells, bind (via Fc receptors) and activate • GBM etc. neutrophils and macrophages • activate complement cascade • damage of the cells, GBM etc.
Type III: Immune-complex-mediated-hypersensitivity Fig. 4 Immunologic reactions after injection of heterologous protein 4
ANTIGEN EXCESS SMALL COMPLEXES (PERSISTENT INFECTION, AUTOIMMUNITY, REPEATED CONTACT WITH ENVIRON- MENTAL ANTIGEN) DEFECT IN SYSTEMS REMOVING IMMUNE COMPLEXES (PHAGOCY- TES & COMPLEMENT) CLEARANCE OF COMPLEXES THEIR DEPOSITION IN TISSUES ACUTE INFLAMMATORY RESPONSE
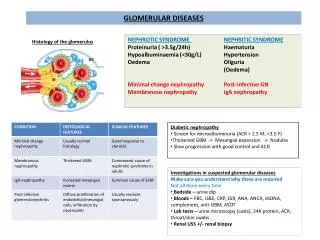
Deposition of complexes may reflect hemodynamic factors (glomeruli) Type IV: Cell-mediated immune injury = delayed-type hypersensitivity Resistant (intracellular) bacterium, foreign tissue etc. activation of TH cells TC, “angry” macrophages, K cells, N(atural) K(iller) cells (=do not require prior immunization) indiscriminate phagocytosis, exudation granulomatous inflammation, contact dermatitis, transplant rejection 22 Pathogenetic mechanisms of glomerular diseases Three typical syndromes: nephritic, nephrotic and chronic glomerulonephritis
Fig. 5 Etiology of glomerular injury - survey 5
Histology • May not correlate with the • clinical presentation • Fig. 6, 7 Various histological • types of glomerulonephritis 6
B: “Minimal changes” GN= lipoid nephrosis: some mesangial proliferation, edematous podocytes, fusion (“loss”) of their foot processes C: Intracapillary mesangial proliferative GN: proliferation of endothelia and mesangium, peeling off of enthelial cells from the GBM, duplication of GBM, “humps” formed by immunocomplexes D: Crescentic GN: proliferation of all components (aggressive white cells, endo- and epithelia, mesangium, epitheloid and giant cells), leakage of fibrin. Hypersensitivity reaction type II or IV E: Membranous GN: Precipitation of immunoglobulins on the outer surface of the GBM (“spikes” complete incorporation of Ig into the membrane) F: Proliferative sclerotizing GN: advanced mesangial proliferation narrowing and destruction of capillaries
221 Visceral epithelial cell (= podocyte) injury Minimal change disease = lipoid nephrosis (Fig. 7 B) 7
Focal segmental glomerulosclerosis (FGS) Focal = 50% of glomeruli are affected by light microscopy Diffuse = 50% affected Segmental = only a part of the glomerular tuft is involved Glomerulosclerosis = obliteration of capillary lumens Nephrotic syndrome. Edematous podocytes, fusion (“loss”) of their foot processes. Unclear podocyte damaging toxin – some lymphokine? React on glucocorticoids
222 Immune complex formation = immune complex disease Glomerulus is highly susceptible to the entrapment or formation of immune complexes Detection: electron microscopy, immunofluorescence (granular appearance) Location of the complexes type of injury and clinical manifestations Fig. 8 Porosity versus permeability 8
2221 Subepithelial deposits Fig. 9 left side 9
Circulating complexes cannost pass through GBM in situimmune complex formation: - circulating cationic antigen, afterwards enter the corresponding antibodies - filtered autoantibody; antigen present in situ (glycoprotein on podocyte cell membrane). CATIONIC ANTIGEN AUTOANTIBODIES POSTINFECTIOUS GN (MOSTLY A, -HEMOLY- TIC STREPTOCOCCI) „ HUMPS“ (FIG. 6C) AMORPHOUS DEPO- SITS MEMBRANOUS NEPHROPATHY. „SPIKES“ (FIG. 6E) SYSTEMIC DISEASES LUPUS NEPHRITIS HEPATITIS B IDIOPATHIC
Injury to the podocytes: membrane attack complex C5b-9 fusion of the foot processes Clinical manifestations of exclusively subepithelial deposits: typically nephrotic. Distortion of slit diaphragms proteinuria Activated complement is not in contact with circulating inflammatory cells lack of inflammatory cell infiltration proteinuria lasts for a long time
Nephrotic syndrome Fig. 10 Pathogenesis of symptoms 10
2222 Subendothelial and mesangial deposits Fig. 9 right side 9
Typically caused by passive entrapment of preformed circulating immune complexes. The nature of the antigen whether subepithelial or subendothelial deposition Lupus nephritis Postinfectious GN: Streptococci, bacterial endocarditis, hepatitis B, malaria Berger Schönlein-Henoch Inflammatory response - Complexes in contact with circulation generate C3a and C5a - Activation of Hageman factor coagulation cascade - Damaged endothelium cytokines and autocoids (local hormones) adhesion molecules activation of endothelial and inflammatory cells
Clinical manifestations: inflammatory and proliferative response typically nephritic syndrome: • active urine sediment: red cells, white cells, cellular and granular casts • GFR • Recovery more rapid, but severe inflammation irreversible cell injury glomerulosclerosis • Hypersensitivity reaction type III
Nephritic diseases (Survey, Fig. 11): 11
Fig. 12 Mechanisms causing reduction of GFR in the nephritic syndrome 12
223 Antibodies directed against GBM antigens Antigen: noncollagenous portion of the 3 chain of type IV collagen Complement and mediators focal glomerular necrosis, crescent formation end-stage renal failure Anti-GBM antibodies bind in a linear pattern to the GBM (without electronoptically dense deposits) Hypersensitivity reaction type II Goodpasture syndrome
Severe damage of capillary wall leaks (rents) in GBM fibrinogen and other plasma components enter Bowman´s space • Crescents= accumulation and proliferation of extracapillary cells • compression of the glomerular tuft rapid renal failure • Crescentic glomerulonephritis (50% glom.) rapidly progressive GN • Etiology: • any severe GN • anti-GBM antibody disease • ANCA-positive disorders • Hypersensitivity reaction type II or IV (?) • 225 Alport syndrome • Congenital defect of collagen
23 Mechanisms of vascular injury • 231 Systemic vasculitis and antineutrophil cytoplasmatic antibodies • Acute systemic process of arteries. • - Large vessel arteritides, e.g. polyarteritis nodosa distal • glomerular ischemia (no inflammation) GFR • - Glomerular tuft, e.g. polyarteritis nodosa, Wegener´s • granulomatosis focal glomerular necrosis, crescents, • active urine sediment • Novel circulating autoantibody – antineutrophil cytoplasmic • antibody = ANCA • Highly specific for systemic necrotizing vasculitides • ANCA respiratory burst of phagocytic cells release of free radicals degranulation injury to endothelial cells
232 Thrombotic microangiopathies Injured endothelial cell loses its natural thromboresistance platelet activation thrombi in the lumen possibly fibrinoid necrosis and fibrin deposition into media Hemolytic-uremic syndrome:thrombocytopenia, microangiopathic hemolytic anemia, renal function Pathogenesis: - verotoxin-producing Escherichia coli damage to endothelia (infantile diarrhea); also immunosuppresives and chemotheraputics - Willebrand factor platelet aggregation; autoantibody against inhibitors of platelet aggregation - antibody-mediated endothelial injury in hyperacute renal transplant rejection
Hematuria, azotemia, hypervolemia. Mild to moderate proteinuria Primary and secondary (=systemic with renal involvement) diseases The majority is of immunologic etiology – hypersensitivity reactions type II or III Local glomerular inflammation breaking filtration membrane porosity, hematuria, proteinuria blocking of glomerular capillaries permeability hypervolemia, uremia Membranous GN (type E) proteinuria, rather slow Proliferating GN (type C and D) hypertension, more acute