Download

1 / 3
30 likes | 44 Views
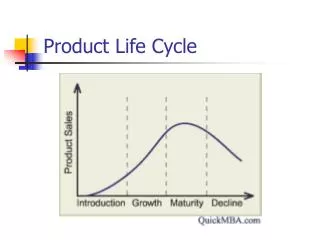
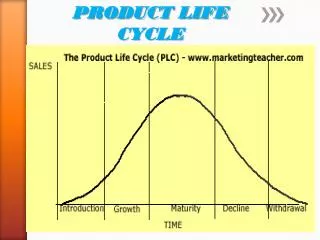
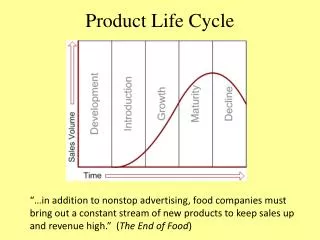
In pharmaceutical industry, product cycle consists of all processes from drug discovery to launch to access, which is closely monitored by regulatory bodies like European Medicines Agency (EMA) for European Union (EU) and Food and Drug Administration (FDA) for United States (US).<br>The various stages include: u2013<br>1.tDiscovery and development.<br>2.tPreclinical research.<br>3.tClinical research with 3 phases.<br>4.tRegulatory body review.<br>5.tPost-authorization / Post-market monitoring.<br><br>To Continue Reading: https://bit.ly/3eK4ztz<br>Contact us;<br>website: https://bit.ly/2W1nV6r <br>Email: sales.cro@pepgra.com<br>

E N D
Post-Authorization Stage of Product Cycle Dr. Nancy Agens, Head, Technical Operations, Pepgra Sales.cro@pepgra.com I. WHAT IS PRODUCT CYCLE In pharmaceutical industry, product cycle consists of all processes from drug discovery to launch to access, which is closely monitored by regulatory bodies like European Medicines Agency (EMA) for European Union (EU) and Food and Drug Administration (FDA) for United States (US). The various stages include: - 1.Discovery and development. 2.Preclinical research. 3.Clinical research with 3 phases. 4.Regulatory body review. 5.Post-authorization monitoring. II. STAGE OF POST-AUTHORIZATION Drug efficacy and safety is studied extensively during clinical trials; however, this is still inadequate and a continued study on the same is needed from the users. This is done by feedback on the same from the end users in the market once the drug is introduced. This decides the life of the drug in the pharmaceutical market. III. UNITED STATES-FOOD AND DRUG ADMINISTRATION (US-FDA) The US-FDA gives detailed guidelines on the various aspects of drug development process including post-market drug safety monitoring. The following aspects are covered: - Supplemental applications – A new application is needed if there is significant difference from the new drug application (NDA) submitted previously. Changes in formulation, dosage must be first approved by FDA. Investigational new drugs (INDs) for marketed drugs- If pharmaceutical company wants approved drug for further use, like different mode of administration or alternative use, it needs to do it under IND. Manufacturer inspections- Planned or surprise inspections and audits are performed by manufacturing plants, both in US and abroad. Inspection may be routine or triggered by a complaint or problem. FDA has authority to shut down a plant if found grossly non-compliant. Drug advertising- FDA regulates drug advertising protocols. Advertising of unapproved products is not allowed. Advertising should be truthful about the performance efficacy and safety of the drug. Promotional labelling, which involves promotional prescribing physicians accompanied information. Generic drugs- Generic manufacturers against originators are not involved in drug research and discovery. They are involved with manufacturing drugs that are already discovered, post expiry of the ten-year patency period. Generic drug should have the same active substance(s) as the reference medicine and is used in the same dose(s) to treat same disease(s). Inactive ingredients need not be the same and so side- effects may differ and that should be mentioned clearly. Generic drugs must conduct only bio-equivalence studies and file abbreviated NDA. Medical device consulting services provide help to develop an FDA of the / Post-market material should prescribing to be by Copyright © 2020 pepgra. All rights reserved 1
drug manufacturers get pre-market approvals. Reporting problems- following channels to enable health professionals, manufacturers, consumers to report problems. E.g. MedWatch is one such gateway for medical products and Medical Product Safety Network (MedSun) is focussed on medical devices. Active surveillance- Under the Sentinel Initiative, to expand and hasten the network of monitoring, FDA will use information from various sources like insurance claims, databases, and registries to add to the post market safety survey process. IV. EUROPEAN UNION European Medicines Agency (EMA) gives elaborate scientific guidelines including pharmacovigilance to pharmaceutical companies products approved in Europe. Post-authorisation (PASS)- Pharmacovigilance assessment committee (PRAC) is responsible for assessing the protocols and results which could be in the form of clinical trials or non-interventional studies. Periodic safety update report (PSUR) are pharmacovigilance documents that provide risk-benefit assessment of medicinal product. requirements of PSUR are elaborated by the EMA information is on risks and benefits is then assessed. Risk management plan (RMP) – This along with good pharmcovigilance practices, provides information to Marketing managers regulatory aspects management lifecycle. Since 2012, all post-authorisation studies (PAS) are required to be published in EU-PAS register and is under European Network of Centres for Pharmacovigilance (ENCePP). Analysing data regarding EMA requested PAS is used in better designing of PAS for future studies. V. CURRENT TRENDS IN EU Primary data capture study pattern is the most used study for risk assessment and effectiveness. Secondary data capture is used for drug utilisation studies. Limitation is that PAS covers only certain types of patient care and not the entire spectrum. Limited data sets and source of data capture is another problem faced in Europe. Primary data collection (data collection specifically for the study) vs secondary (where data is already collected for another purpose) is the methodology of choice that is gaining popularity as confounding factors are fewer. Engel and others studied PASS protocols and assessments during 2012 to 2015 from PRAC minutes, EMA and ENCePP. They recommended to increase the availability of data from PASS and EMA to the EU-PAS register. Reviewing protocols and PRAC comments should sensitise PASS stakeholders to put more thought into making protocols that are in line with pharmacodynamics, and guidelines of regulatory body. Farcas and others studies the risk minimization measures (RMMs) from the EU-PAS register effectiveness. RMMs are related to safety-related outcomes and their effectiveness is important to ensure safety of the user. Also, their alignment to the Good Practices guidelines recommendations was assessed along with process and outcome indicators. Pharmacoepidemiology and FDA has from ENCePP and health record and regulatory for those safety studies risk Submission pharmacokinetics, and cumulative and their about the risk regarding Pharmacovigilance They Copyright © 2020 pepgra. All rights reserved 2
recommended stricter adherence to the recommendations established endpoints for evaluation of RMM effectiveness. to achieve VI. ADDITIONAL GUIDELINES FOR SPECIALISED AREAS Medical device regulatory affairs provide different specialised areas paediatric medicine, medicines. For e.g. in case of orphan medicines that are drugs used in the treatment of rare conditions like inherited errors of metabolism, PASS is important and quick access to this information is important for effective treatment. The significance of post- approval drug studies lies in the understanding that true performance and safety of a drug can only be understood once it is introduced in the population and used under various circumstances. It is important to accurately record these events and submit to the concerned registries to be compiled, analysed, and interpreted. At the same time, ready access to this data is needed to understand risk-benefits and make an informed decision. The outcomes of studies act as inputs to analyse study protocols and this continuous process of feedback is needed to have a robust system. guidelines like and for oncology, orphan help physicians Copyright © 2020 pepgra. All rights reserved 2