Download

1 / 12
120 likes | 354 Views
Scoring Matrices. April 23, 2009 Learning objectives- Last word on Global Alignment Understand how the Smith-Waterman algorithm can be applied to perform local alignment. Have a general understanding about PAM and BLOSUM scoring matrices. Homework 3 and 4 due today Quiz 1 today

E N D
Scoring Matrices • April 23, 2009 • Learning objectives- • Last word on Global Alignment • Understand how the Smith-Waterman algorithm can be applied to perform local alignment. • Have a general understanding about PAM and BLOSUM scoring matrices. • Homework 3 and 4 due today • Quiz 1 today • Writing topic due today • Homework 5 due Thursday, April 30.
Global Alignment output file Global: HBA_HUMAN vs HBB_HUMAN Score: 290.50 HBA_HUMAN 1 VLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFP 44 |:| :|: | | |||| : | | ||| |: : :| |: :| HBB_HUMAN 1 VHLTPEEKSAVTALWGKV..NVDEVGGEALGRLLVVYPWTQRFFE 43 HBA_HUMAN 45 HF.DLS.....HGSAQVKGHGKKVADALTNAVAHVDDMPNALSAL 83 | ||| |: :|| ||||| | :: :||:|:: : | HBB_HUMAN 44 SFGDLSTPDAVMGNPKVKAHGKKVLGAFSDGLAHLDNLKGTFATL 88 HBA_HUMAN 84 SDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTPAVHASLDKF 128 |:|| || ||| ||:|| : |: || | |||| | |: | HBB_HUMAN 89 SELHCDKLHVDPENFRLLGNVLVCVLAHHFGKEFTPPVQAAYQKV 133 HBA_HUMAN 129 LASVSTVLTSKYR 141 :| |: | || HBB_HUMAN 134 VAGVANALAHKYH 146 %id = 45.32 %similarity = 63.31 (88/139 *100) Overall %id = 43.15; Overall %similarity = 60.27 (88/146 *100)
Smith-Waterman Algorithm Advances inApplied Mathematics, 2:482-489 (1981) Smith-Waterman algorithm –can be used for local alignment -Memory intensive -Common searching programs such as BLAST use SW algorithm
Smith-Waterman (cont. 1) a. Initializes edges of the matrix with zeros b. It searches for sequence matches. c. Assigns a score to each pair of amino acids -uses similarity scores -uses positive scores for related residues -uses negative scores for substitutions and gaps d. Scores are summed for placement into Mi,j. If any sum result is below 0, a 0 is placed into Mi,j. e. Backtracing begins at the maximum value found anywhere in the matrix. f. Backtrace continues until the it meets an Mi,j value of 0.
Smith-Waterman (cont. 2) H E A G A W G H E E Put zeros on top row and left column. Assign initial scores based on a scoring matrix. Calculate new scores based on adjacent cell scores. If sum is less than zero or equal to zero begin new scoring with next cell. P A W H E A E 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 5 0 5 0 0 0 0 0 0 0 0 0 3 0 2012 4 0 0 0 10 2 0 0 1 12182214 6 0 2 16 8 0 0 4101828 20 0 0 82113 5 0 41020 27 0 0 6131912 4 0 416 26 This example uses the BLOSUM45 Scoring Matrix with a gap penalty of -8.
Smith-Waterman (cont. 3) H E A G A W G H E E P A W H E A E 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 5 0 5 0 0 0 0 0 0 0 0 0 3 0 2012 4 0 0 0 10 2 0 0 1 12182214 6 0 2 16 8 0 0 4101828 20 0 0 82113 5 0 41020 27 0 0 6131912 4 0 416 26 Begin backtrace at the maximum value found anywhere on the matrix. Continue the backtrace until score falls to zero AWGHE || || AW-HE Score=28
Calculation of similarity score and percent similarity A W G H E A W - H E 5 15 -8 10 6 Blosum45 SCORES GAP PENALTY (novel) % SIMILARITY = NUMBER OF POS. SCORES DIVIDED BY NUMBER OF AAs IN REGION x 100 Similarity Score= 28 % SIMILARITY = 4/5 x 100 = 80%
Why search sequence databases? 1. I have just sequenced something. What is known about the thing I sequenced? 2. I have a unique sequence. Does it have similarity to another gene of known function? 3. I found a new protein sequence in a lower organism. Is it similar to a protein from another species?
Perfect searches for similar sequences in a database • First “hit” should be an exact match. • Next “hits” should contain all of the genes that are related to your gene (homologs). • Next “hits” should be similar but are not homologs
How does one achieve the “perfect search”? • Consider the following: • Scoring Matrices (PAM vs. BLOSUM) • Local alignment algorithm • Database • Search Parameters • Expect Value-change threshold for score reporting • Translation-of DNA sequence into protein • Filtering-remove repeat sequences
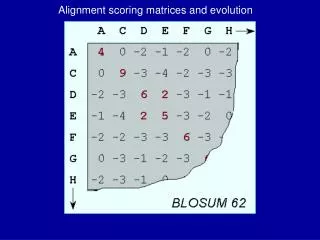
PAM-1 BLOSUM-100 Small evolutionary distance High identity within short sequences Which Scoring Matrix to use? PAM-250 BLOSUM-20 • Large evolutionary distance • Low identity within long sequences
BLOSUM Scoring Matrices Which BLOSUM Matrix to use? BLOSUM Identity (up to) 80 80% 62 62% (usually default value) 35 35% If you are comparing sequences that are very similar, use BLOSUM 80. Sequences that are more divergent (dissimilar) than 20% are given very low scores in this matrix.