Download

1 / 66
660 likes | 679 Views
This document discusses the evaluation of a new software tool (Structure 2.0) for association mapping in population genetics. It covers the principles, methods, and advantages of using Bayesian inference and Markov Chain Monte Carlo (MCMC) techniques for population structure analysis. The text also explores strategies for grouping individuals and detecting admixture complications. Moreover, it presents detailed information on running the MCMC program and assessing parameters for population genetics studies.

E N D
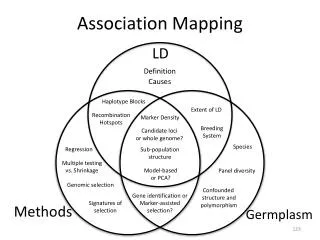
Evaluation of a new tool for usein association mapping Structure Reinhard Simon, 2002/10/29
Software Structure 2.0http://pritch.bsd.uchicago.eduPritchard JK, Stephens M, Donelly P (2000):Inference of population structure using multilocus genotype data.Genetics, 155: 945-959
Associations – the ideal Cases Controls
Test for association A diploid locus: Pearsons Chi-square test
Associations – the less ideal Cases Controls
Associations – simple admixture Cases Controls
Associations – admixture complications Cases Controls
Associations – admixture complications Cases Controls High frequency of associated loci may indicate problems with underlying population structure (=stratification).
Associations – accounted for Cases Controls
Questions • Is there a stratification? • If so: - how many subpopulations - which individual belongs to which subpopulation
Test for stratification - principle Summarizing over all loci: • Xi is Chi-square at i-th locus • Null hypothesis: no differences between allele frequencies over all loci • df equal to sum of df at individual locus Pritchard: 1999
Test for stratification – ctd. Observations: • strong positive selection requires increase of #loci • subgroup specific markers decrease number of necessary loci Pritchard: 1999
How to group individuals? • Based on distance measures • Based on models
Pair wise distance measures Jaccard Nei & Li Sokal & Michener
Model based Bayesian inference • Bayesean statistics: Uncertainty is modeled using probabilities • probability statements are made about model parameters Advantages: • very general framework • assumptions are made explicit and are quantified
Bayesian inference – how? • Bayesian inference centers on the posterior distribution p(theta|X), e.g.a genetic model of the distribution of allele frequencies • However, analytic evaluation is seldom possible ....
Bayesian inference - methods Alternatives: • Numerical evaluation • approximation • simulation, e.g. Markov Chain Monte Carlo Methods
Simulation methods for Bayesian inference - general • Generate random samples from a probability distribution (e.g. normal) • Construct histogram • If sample is large enough, this allows to calculate mean, variance, ... • MCMC allows to generate large samples from any probability distribution
Markov Chain behaviour • Reaches an equilibrium (basic MCMC theorem) and • the present state depends only on the preceding: “The future depends on the past only through the present.”
MCMC - strengths • freedom in inference (e.g. simultaneous estimation, estimation of arbitrary functions of model parameters like ranks or threshold exceedence) • Coherently integrates uncertainty • Only available method for complex problems
MCMC – contra • computational intensive • requires often specialized software
Inferring population structure X = genotypes of sampled invidualsunknown:Z = population of originP = allele frequencies in all populationsQ = proportion of genome that originates from population k Pr(Z, P, Q|X) ~ Pr(Z) * Pr(P) * Pr(Q) * Pr(X|Z,P,Q) Solution:Using MCMC for Bayesian inference;simultaneous estimation of Q, Z and P.
Basic MCMC algorithm – no admixture (Q) Initialize:Random values for Z (pop), e.g. from Pr(z) = 1/k Repeat for m=1,2,...1. Sample P(m) from Pr(P|X, Z(m-1) (estimate allele frequencies) 2. Sample Z(m) from Pr(Z|X, P(m)) (estimate population of origin for each indiv.)
Basic MCMC algorithm – with admixture (Q) Initialize:Random values for Z (pop), e.g. from Pr(z) = 1/k Repeat for m=1,2,...1. Sample P(m), Q(m) from Pr(P, Q|X, Z(m-1) (estimate allele frequencies) 2. Sample Z(m) from Pr(Z|X, P(m), Q(m)) 3. Update alpha (admixture proportion)
Program – data types • marker: SNP, microsatellites AFLP, RFLP, ... (biallelic) • ploidy: >1 • extra optional information for inclusion: • prior knowledge on groups (e.g. geographic location) • genetic map location of marker
Example – S.t. tuberosum vs andigena Other:1st 30 genotypes from tuberosum 2nd 20 genotypes from andigena
Example – S.t. tuberosum vs andigena PNA: Estimation of k Simulation # k Pr(k)
Example – S.t. tuberosum vs andigena PNA: assignment 1 = tbr; 2 = adggenotypes #31-#3: adg from Indiagenotype #49: adg from Ecuador
Example – S.t. tuberosum vs andigena Parameter change: allow admixture Ancestry Model Info Use Admixture Model * Infer Alpha * Initial Value of ALPHA (Dirichlet Parameter for Degree of Admixture): 1.0 * Use Same Alpha for all Populations * Use a Uniform Prior for Alpha ** Maximum Value for Alpha: 10.0 ** SD of Proposal for Updating Alpha: 0.025Frequency Model Info Allele Frequencies are Independent among Pops * Infer LAMBDA ** Use a Uniform Lambda for All Population ** Initial Value of Lambda: 1.0
Example – S.t. tuberosum vs andigena Parameter change: allow admixture
Example – S.t. tuberosum vs andigena Parameter change: allow admixture
Example – S.t. tuberosum vs andigena Parameter change: allow admixture
Example – andigena K = 2
Example – andigena K = 3
Example – andigena K = 3