Download
1 / 1
10 likes | 163 Views
40. 20. 0. -20. -40. -60. -40. -45. -50. -55. -60. Solid-state 31 P and 13 C NMR of extremely bulky phosphines. Ren é T. Boeré , Paul Hazendonk and Dinu Iuga Department of Chemistry and Biochemistry and Department of Physics, The University of Lethbridge, Alberta, Canada.

E N D
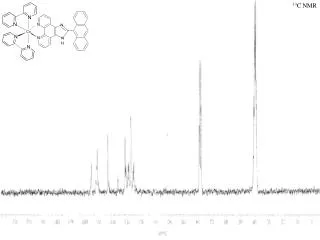
40 20 0 -20 -40 -60 -40 -45 -50 -55 -60 Solid-state 31P and 13C NMR of extremely bulky phosphines René T. Boeré, Paul Hazendonk and Dinu Iuga Department of Chemistry and Biochemistry and Department of Physics, The University of Lethbridge, Alberta, Canada Introduction Solid-state 13C NMR results Solid-state 31P NMR Results Figure 3: MAS 13C NMR spectra of the solid phosphines Figure 2: MAS 31P NMR spectra of the solid phosphines Triaryl phosphines are among the most common ligands in coordination chemistry and are of immense importance in all branches of homogeneous catalysis. They are also important reagents in organic and inorganic chemistry. A major theme in the coordination chemistry of phosphines has been their steric bulk.1 The other major theme has been the basicity of the phosphines, and in particular the way in which the three organic substituents at phosphorus can modify this property. We have recently synthesized and structurally characterized a series of phosphines of differing steric bulk and basicity incorporating the extremely bulky 2,6-diisopropyl group, Dipp.2 Indeed, the substitution of three aryl groups each bearing two ortho isopropyl groups represent the currently most bulky phosphines ever to be synthesized. The closely related Dipp3P prepared in our group and Tripp3P prepared by Yoshifuji and Sasaki are the world’s bulkiest phosphines.3 31P NMR spectra of Dipp3P in the solid-state (MAS) -52.7 31P NMR spectra of Trip3P in the solid-state (MAS) -51.9 1H->13C CP spectra of Dipp3P 6 kHz 4 kHz 0 kHz -48.5 nMAS=12kHz 1H->13C CP spectrum of Tripp3P nMAS=6kHz 150 140 130 120 110 30 20 ppm nMAS=3kHz Crystallography 1H->13C CP spectra of Dipp2PPh Static This CH3 signal is shifted to lower frequency due to increased diamagnetic shielding (see circled atom in the diagram of the crystal structure). 160 140 120 35.5 25 ppm Figure 1: Molecular structures as found in the crystal structures (H atoms omitted) 0 0 0 -50 -50 -50 -100 -100 ppm 31C isotropic peaks of DippPPh2 31P spectra of Dip2PPh 31P NMR spectra of DippPPh2 in the solid-state (MAS) 160 150 140 130 120 40 30 20 ppm 25 kHz (XY8 1H dec.) 25 kHz (cw 1H dec.) 12 kHz 3 kHz 0 kHz -20.7 Very high resolution 13C NMR spectra were obtained for all four compounds. The CH3 environs of the isopropyl groups lead to four distinct methyl carbon shifts in both Dipp3P and DippPPh2. These compounds differ only in specific details of their other chemical shifts. However, both Tripp3P and Dipp2PPh display more spectral lines than expected in each region of the spectrum. In the latter, this is easily explained by the low-symmetry environment and the presence of two different molecules in the unit cell. For Tripp3P, the para CH3 confuse the spectra, leading to many overlapping methyl C lines (there are 18 different CH3 environments!) nMAS=12kHz -31.5 ppm -28.5 ppm -32.7 ppm 33.5 ppm nMAS=5kHz Dipp3P Tripp3P -20.8 ppm nMAS=3kHz 50 0 -50 -100 ppm References The sharpest MAS signal belongs to Tripp3P, which also has a cylindrical shielding tensor in the static spectrum that is narrow (σ≈ σ). Similar shielding tensors were reported previously for other symmetrical ortho substituted triarlyphosphines.4 By contrast, Dipp3P which crystallizes in a higher symmetry space group displays considerably broader lines, and in particular in the static spectrum appears as two different powder-patterns of differing intensity but similar breadth. This seems to reflect twinning by merohedry which was detected in all crystals measured by Dipp3P no matter what solvent was used for the recrystallization. The twinning occupancy refined in the single-crystal model to ≈16%. Both of the mixed Dipp/Ph phosphines display more typical spectra for triaryl phosphines. However, Dipp2PPh also displays the obvious presence of the two distinct kinds of molecules found in its lattice. These correspond to the peaks centred at -31.5 and -32.7 ppm. On of the two components is broadened compared to the other. This mixture of the two environments persists into the static spectrum. While the two molecular structures are virtually superimposable in the two sites, there is a small difference in the twist angle of the unsubstituted rings which may account for this difference. The spectrum of DippPPh2 has a distinctly asymmetric shielding tensor in the static spectrum, reflecting the very different positions of the three aryl rings which are twisted 12, 52 and 61° from the three-fold axes of the molecules. • (a) Tolman, C. A. Chem. Rev. 1977, 77, 313-348. (b) Tolman, C. A. J. Am. Chem. Soc. 1970, 92, 2956-2965. (c) Andersen, N. G.; Keay, B. A. Chem. Rev. 2001, 101, 997-1030. . • Boeré, R. T.; Zhang, Y. J. Organomet. Chem. 2005, 690, 2651-2657.. • Sasaki, S.; Sutoh, K.; Murakami, M.; Yoshifuji, M. J. Am. Chem. Soc.2002, 124, 14830-14831. • Penner, G. H.; Wasylishin, R. E. Can. J. Chem.1989, 67, 1909-1913. Dipp2PPh DippPPh2 The solid-state structures of all four phosphines examined in this study have been determined by single-crystal X-ray diffraction. Each have distinct structures that are directly reflected in the solid-state 31P NMR spectra (shown at right). Thus: Dipp3P R3 twinning by merohedry to R322; Z = 3 (one molecule per equiv. position so that the molecule must have a three-fold axis of symmetry). Tripp3P , ordered structure; Z = 2 (one molecule per equiv. position, no symmetry applied). Dipp2PPh P21/n; Z = 8 (two independent molecules per equiv. position; so symmetry applied.) DippPPh2Pbca; Z = 8 (one molecule per equiv. position, no symmetry applied). Acknowledgements We thank NSERC-Canada and the University of Lethbridge for funding. The Alberta Proteomics Network and WCED supported the acquisition of the spectrometer. Twyla Gietz, Jason Masuda, Sonja Seagrave and Yuankui Zhang prepared the compounds and Masood Parvez performed some of the X-ray crystallographic studies.