Download

1 / 38
390 likes | 880 Views
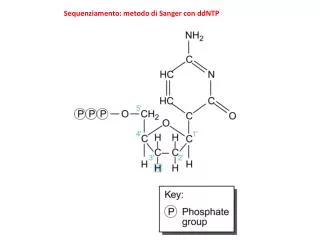
Il sequenziamento genico. Struttura del DNA. Doppia elica formata da due filamenti di ac. nucleico uniti da ponti ad idrogeno Ac. nucleico: catena di nucleotidi Nucleotide: nucleoside + ac. fosforico Nucleoside: desossiribosio + base azotata Basi azotate: adenina, citosina, guanina e timina.

E N D
Struttura del DNA • Doppia elica formata da due filamenti di ac. nucleico uniti da ponti ad idrogeno • Ac. nucleico: catena di nucleotidi • Nucleotide: nucleoside + ac. fosforico • Nucleoside: desossiribosio + base azotata • Basi azotate: adenina, citosina, guanina e timina
Le basi azotate • Unico elemento variabile all’interno dell’ac. nucleico • Sequenziamento: individuazione della successione delle basi azotate nel filamento di ac. nucleico
Le basi azotate • Complementarità fra i due filamenti: • A-T • G-C • 5’-3’: forward • 3’-5’: reverse
Prima del sequenziamento • Scelta del bersaglio • Presente in tutti gli organismi • Conservato ma contenente regioni variabili • Disponibilità di un database contenente le sequenze con cui si vuol confrontare il bersaglio • Scelta dei primer • Estrazione del DNA dalle cellule
Le fasi del sequenziamento • Amplificazione del bersaglio • Verifica dell’amplificato • Purificazione • Verifica • PCR di sequenziamento • Purificazione dell’amplificato
Miscela di sequenziamento • Primer • Due amplificazioni parallele usando in ciascuna un solo primer (per i filamento forward o reverse) • Tampone • Polimerasi • Nucleotidi • Nucleotidi terminator
Nucleotidi terminator • Nucleotide (A) che blocca l’allungamento del filamento di ac. nucleico poiché la mancanza del gruppo ossidrile in 3’ impedisce l’attacco all’ac. fosforico del nucleotide successivo
PCR di sequenziamento (metodo manuale) • Si esegue in quattro provette diverse • Tutte e quattro le provette contengono: polimerasi, nucleotidi normali e primer • Ciascuna delle quattro provette contiene inoltre un diverso tipo di nucleotide terminator (adenina-terminator, guanina-terminator, . . .) • Annealing del primer • Allungamento del primer: • Legame di un nucleotide normale: l’allungamento prosegue • Legame di un nucleotide terminator: l’allungamento si blocca • In ciascuna delle 4 provette contenente nucleotidi terminator con una diversa base azotata si formano soltanto filamenti che terminano con tale base. Tali filamenti avranno tutte le lunghezze possibili a seconda che il nucleotide terminator si sia legato alla 1°, 2°, . . , ennesima, posizione possibile
Il principio del sequenziamento (metodo manuale) • Si esegue una elettroforesi disponendo i prodotti di amplificazione delle 4 provette in 4 corsie parallele così che in ognuna di esse siano presenti filamenti che terminano con una diversa base • Si usa un gel ad altissima risoluzione, capace di separare frammenti di DNA che differiscono fra loro per un solo nucleotide. Il gel (0,3-0,4 mm di spessore) è interposto fra due lastre di vetro di notevole lunghezza (40 cm circa ).
Il principio del sequenziamento (metodo manuale) • La migrazione è inversamente proporzionale alla lunghezza del filamento • La posizione della banda indica la posizione occupata all’interno della sequenza, mentre il tipo di base è indicato dalla corsia in cui tale banda si trova A T A C A G C T G T T . . . . . .
Nucleotidi terminator marcati(metodo automatico) • I nucleotidi terminator sono marcati con un fluorocromo • Si usa un fluorocromo diverso per ciascuno dei quattro tipi di terminator
PCR di sequenziamento (metodo automatico) • Si esegue in una singola provetta • Tutte le provette contengono: polimerasi, nucleotidi normali, nucleotidi terminator e primer • I nucleotidi terminator sono marcati con fluorocromi • Annealing del primer • Allungamento del primer: • Legame di un nucleotide normale: l’allungamento prosegue • Legame di un nucleotide terminator: l’allungamento si blocca • Nella provetta si formano contemporaneamente filamenti che avranno tutte le lunghezze possibili a seconda che il nucleotide terminator si sia legato alla 1°, 2°, . . , ennesima, posizione della sequenza. Si avranno quindi contemporaneamente filamenti terminanti con ciascuna delle quattro basi
Il prodotto della PCR (sequenziamento automatico) • Se il bersaglio è composto da n nucleotidi si avranno filamenti di tutte le lunghezze possibili, da 1 ad n • Indipendentemente dalla lunghezza, tutti i filamenti terminanti con adenina emetteranno fluorescenza di tipo A ; quelli terminanti con citosina, di tipo B; quelli terminanti con guanina, di tipo C, e quelli terminanti con timina, di tipo D
Il sequenziatore automatico • E’ un apparecchio che, dopo aver prelevato il prodotto della PCR di sequenziamento, lo sottopone ad elettroforesi all’interno di un capillare • Un raggio laser colpisce il capillare eccitando la fluorescenza dei fluorocromi che lo attraversano • Un cellula fotoelettrica rileva i segnali fluorescenti che vengono memorizzati
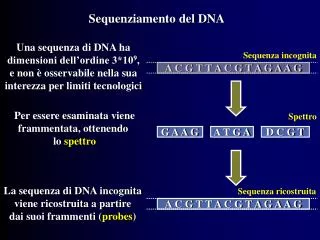
L’elettroferogramma • Durante l’attraversamento del capillare i vari spezzoni di DNA vengono colpiti da un raggio laser • Il raggio laser eccita i vari fluorocromi che marcano i singoli spezzoni • Ciascuno dei quattro fluorocromi emette una diversa lunghezza d’onda • Una cellula fotoelettrica rileva sequenza, tipo ed intensità delle varie emissioni luminose ed il tutto viene registrato in forma grafica • La sequenza dei picchi corrisponde alla sequenza dei nucleotidi; il tipo (colore del picco) corrisponde al tipo di base azotata mentre l’intensità (altezza del picco) è irrilevante
L’elettroferogramma • Normalmente il sistema interpreta automaticamente l’elettroferogramma • Quando l’interpretazione non è ovvia, il sistema inserisce una N al posto della base azotata mancante, questa può essere corretta manualmente, dopo aver letto ad occhio l’elettroferogramma, o in base alla sequenza del filamento reverse G
Il sequenziamento in microbiologia • Identificazione • Sequenziamento di regioni specie-specifiche • Antibiogramma • Sequenziamento di regioni in cui possono aversi mutazioni associate alla resistenza ai farmaci • Filogenesi • Confronto della sequenza della medesima regione in specie diverse, per ricostruirne la storia evolutiva
Identificazione mediante sequenziamento • Scelta della regione da sequenziare • 16S rDNA • Internal transcribed spacer • 23S rDNA • hsp65 • rpoB • Sequenziamento • Confronto della sequenza con quelle presenti in un database
GenBank • Banca dati pubblica (in Internet) contenente circa 50 milioni di sequenze geniche delle più varie regioni di, praticamente, tutti gli organismi viventi • Chiunque può depositare (ovviamente non in maniera anonima) sequenze in GenBank • E’ possibile confrontare la propria sequenza con tutte quelle presenti in GenBank • Un motore di ricerca individua e restituisce le sequenze presenti in GenBank che hanno il più elevato grado di somiglianza con la sequenza in esame
Interpretazione • Identità 100% con una specie nota • Identità 100% con una sequenza non appartenente a specie conosciute • Identità <100% con una specie nota (il significato varia a seconda della regione in esame) • Verifica delle discordanze • Correzione della sequenza (identità 100%) • Nuovo sequevar • Nuova specie
Antibiogramma mediante sequenziamento • Scelta della regione da sequenziare • rpoB • katG • embB • pncA • Sequenziamento • Confronto con la sequenza wild type
Interpretazione • Confronto con la sequenza wild type • Identità 100% = sensibilità • Mutazioni = resistenza • Conferma con l’antibiogramma fenotipico
Le mutazioni • Principali mutazioni • Inserzione • Delezione • Sostituzione • Conservativa • Non conservativa • Mutazioni: errori della natura? • La selezione naturale premia le mutazioni favorevoli e elimina quelle sfavorevoli • Regioni genetiche più o meno tolleranti
Mutazioni e filogenesi • Evoluzione da un organismo ancestrale • Sviluppo di nuovi organismi per effetto di mutazioni e selezione naturale • Il numero, il tipo e la posizione delle mutazioni comparse, in regioni conservate, durante l’evoluzione costituiscono il metro con cui è possibile misurare le distanze filogenetiche • Numero di mutazioni tanto più elevato quanto più remoto è il progenitore ancestrale • Organismi con mutazioni concatenate appartengono allo stesso phylum
Regioni di studio • Ideale: l’intero genoma • Compromesso accettabile:16S rRNA • Sequenziamento • Multiallineamento • Costruzione dell’albero filogenetico
L’albero filogenetico 4 mutazioni 5 mutazioni 5 mutazioni 7 mutazioni 4 mutazioni 2 mutazioni
L’albero filogenetico 4 mutazioni 5 mutazioni 5 mutazioni 7 mutazioni 4 mutazioni 2 mutazioni
L’albero filogenetico, limiti • Non sempre, alberi basati su sequenze di regioni diverse, sono compatibili • Non sempre alberi basati sulle stesse sequenze, ma costruiti con algoritmi diversi, sono sovrapponibili
Conclusioni • L’uso del sequenziamento per l’identificazione e l’antibiogramma è particolarmente vantaggioso con: • Organismi a crescita lenta • Organismi difficilmente coltivabili • Organismi non coltivabili • Organismi morti (paleomicrobiologia) • Per l’identificazione il sequenziamento è il metodo di riferimento; NON lo è per l’antibiogramma • Il sequenziamento è una metodica: • Rapida • Automatizzata • Economica