Download

1 / 38
480 likes | 1.09k Views
Lecture 6. Molecular cloning. With thanks to David Tscharke @ RSB. Lecture overview. AKA gene cloning or DNA cloning The generation of identical copies of a piece of DNA Propagated in bacteria that originate from a single cell Was the first recombinant DNA technology

E N D
Lecture 6 Molecular cloning With thanks to David Tscharke @ RSB
Lecture overview • AKA gene cloning or DNA cloning • The generation of identical copies of a piece of DNA • Propagated in bacteria that originate from a single cell • Was the first recombinant DNA technology • Joining of DNA from one species to another such that both are propagated biologically • Molecular Meccano, Molecular Lego… • Brings together a number of techniques in molecular biology • Nothing to do with reproductive cloning!
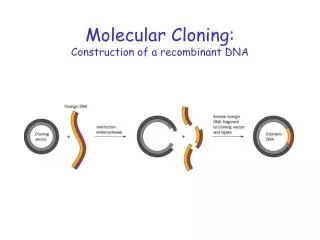
Cloning DNA into plasmids Bacterial culture Extract plasmid Cut DNA with restriction enzymes - generate cohesive ends In vitro In vivo + ligase “Insert” Propagate in bacterial culture transformation
Why clone in plasmids and bacteria Amplification up to 200x Establish and maintain clonal purity Amplification x billions!
Why clone in plasmids and bacteria Amplification in culture Modify extract Express protein Move to new vector Extract product Determine sequence Transfer to: - eukaryotic cells - plants - fungi - virus - etc… Express in vitro
Cloningversus sub-cloning Cloning is when the source of the ‘insert’ DNA is from genomic DNA, cDNA or another non-plasmid source RE cut Or PCR RE cut
Cloning versussub-cloning Subcloning is when the source of the ‘insert’ DNA is from another plasmid RE cut Or PCR RE cut
Cloning is a marriage of techniques Cloning requires • Easy to extract and manipulate DNA ‘vector’ or host • Plasmid • Bacteriophage • Easy to extract and manipulate DNA ‘vector’ or host • Plasmid • Bacteriophage • Tools for cutting and joining DNA • Restriction enzymes • Ligase • Ability to move the new DNA back in vivo • Transformation • Selection and screening • Ability to identify clones of bacteria harbouring the desired recombinant plasmid • Colony PCR and sequencing
The essentials for a cloning vector Unique restriction sites PstI BamHI EcoRI Antibiotic resistance Origin of replication
The essentials for an overexpression vector Ribosome binding site = Shine-Dalgarno sequence Gene of interest promoter terminator Antibiotic resistance Origin of replication
Antibiotic resistance For selection of bacteria containing the plasmid Antibiotic Target Mechanism of of action resistance ampicillin murein-layer b-lactamase bacterial cell wall kanamycin ribosomal 30S aminophosphotransferase subunit tetracyclin ribosomal 30S efflux (pump) subunit chloramphenicol ribosomal 50S chloramphenicol subunit acetyltransferase
Antibiotics and mechanism of resistance ampicillin b-lactamase kanamycin APHs phosphorylate
Antibiotics and mechanism of resistance Exported by efflux pump (transmembrane protein) tetracyclin One or both OH groups acetylated by chloramphenicol acetyltransferase (CAT) chloramphenicol
Footnote to penicillin resistance b-lactamase acts in the periplasmic space and is secreted into the culture medium • Cells without resistance gene can survive in the neighborhood • Problem occurs if • ampR is on high copy number plasmid • cells are allowed to grow for a long time (“stationary phase”) “Satellites”
Multiple cloning site • Cluster of unique restriction enzyme recognition sites • used to insert foreign DNA • Abbreviated to MCS, also called a polylinker 23 restriction enzyme sites shown
Summary • Essential elements of a plasmid cloning vector • Ori – origin of replication • e.g. ColE1 origin • Selection marker – antibiotic resistance • At least one unique restriction site • A deluxe plasmid cloning vector has: • Restriction sites clustered in an MCS
Cloning is a marriage of techniques Cloning requires • Easy to extract and manipulate DNA ‘vector’ or host • Plasmid • Bacteriophage • Tools for cutting and joining DNA • Restriction enzymes • Ligase • Ability to move the new DNA back in vivo • Transformation • Selection and screening • Ability to identify clones of bacteria harbouring the desired recombinant plasmid • Colony PCR and sequencing
You need an enzyme to cut DNA EcoRI 5’ GAATTC G AATTC 3’ CTTAAG CTTAA G 5’ overhang 5’ protruding 5’ sticky SmaI 5’ CCCGGG CCC GGG 3’ GGGCCC GGG CCC blunt PstI 3’ overhang 3’ protruding 3’ sticky 5’ CTGCAG CTGCA G 3’ GACGTC G ACGTC Ends can be re-joined by ligase if the 5’ phosphate is intact
Ligase Ligase • Ligase repairs broken • phosphodiester bonds • Uses ATP (one for each bond repaired) • Most common enzyme for joining DNA in vitro Lodish Fig. 9-11
Ends that can be re-joined by ligase • Any blunt end to any other blunt end • Sticky ends as long as they are ‘cohesive’ • Overhanging bases must be complementary • Ligated ends can be cut again if the recognition site is regenerated
Vector and insert must be cut with enzymes that produce ends compatible for ligation Let’s play the ligation game!
cut fragments out Purify (silica) Often need to purify the cut DNA Preparative gel electrophoresis Several wells joined Restriction digest +
Temperature problem for ligation • The problem • Association of DNA ends is best at low temperature • Also hybridisation of short sticky ends • But ligase works best at 37 oC • Solutions include • Long incubations at 16 oC (overnight) • Molecular ‘crowding agents’ and loads of ligase • Polyethylene glycol (PEG), long chain polymer • 10x more ligase
Summary Joining DNA in vitro Digestion of plasmid DNA with restriction enzymes Isolation of target DNA and digestion to generate compatible ends Often gel purification of fragments Ligation of the two DNA molecules It’s trickier than it might appear at first
Cloning should be this simple + ligase • Also need: • T7 promoter and RBS before gene of interest • T7 terminator after gene of interest
NdeI EcoRI 2 different restriction sites Plasmid and insert must be cut with same restriction enzymes Ends not cohesive • Plasmid re-ligates only with insert • Plasmid cannot ligate with plasmid • Insert cannot ligate with insert In the plasmid: 5’ CATATG CA 3’ GTATAC GTAT NdeI 5’ GAATTC AATTC 3’ CTTAAG G EcoRI
What if… • The insert has internal NdeI or EcoRI sites • Use different restriction sites • taking care that a RBS is at the right place…. • Order a synthetic gene • only $0.5 per base pair • Use a restriction-free method
Cloning methods for the 21st century • Ligations can be tricky • Needs matching overhang • Ligase needs phosphorylated 5’ ends • blunt-ended ligations (i.e. no overhang) 10-fold less effective • Restriction-free methods • 20 nucleotide overlaps are better than 1-4 nucleotide overlaps • - But no restriction enzyme delivers large overlaps • SLIC “sequence and ligation independent cloning” • CPEC “circular polymerase extension cloning” • SLiCE “seamless ligation cloning extract”
T4 DNA pol 3’ 5’ SLIC Appl. Eviron. Microbiol. 2012 doi:10.1128/AEM.00844-12 • Linearize vector • Cut with restriction enzyme • Combine linearized vector and insert • Add T4 DNA polymerase (2.5 min at room temperature) • Put on ice to stop digestion • 10 min annealing (on ice) • Transform cells Polymerase activity Exonuclease activity 3’ 5’
SLIC – why it works • T4 Pol exonuclease generates 5’ overhangs in plasmid and insert • Ends must be complementary for about 20 nucleotides • Spontaneous hybridisation • Don’t worry about 5’ phosphorylation • No ligase • Segments of ssDNA acceptable • The polymerases, kinases and ligases present in E. coli • do the rest to heal the vector! Potential problem Annealing at low temperature can lead to mis-hybridization
CPEC Nat. Protoc. 6, 242 (2011) • Use the strands from the insert as primers • “Overlap extension PCR” Denatured insert Denatured linearized vector • Melting temperatures of all overlapping regions must have similar Tm • Tm between 60 and 70 oC for stringent hybridization • 15 – 35 bases overlap
CPEC • Anneal • One round of overlap extension PCR • Transform cells • E. coli heals the nicks in the vector • Potential problem • Vector copy made by PCR • Even high-fidelity polymerases introduce ~1 error per 9000 bp
A vector can be linearized by PCR • A single round of PCR makes a linear copy • Use high-fidelity polymerase (e.g. Phusion) Errors do not propagate in a single round Primers designed to introduce overlapping regions with the insert
SLiCE Nucl. Acids Res. 40, e55 (2012) • recombination system • Needs 20 nucleotide overlap • Ligates insert into vector even if the • vector has some non-matching overhangs • Express necessary enzymes in a special • E. coli strain and use cell-extract • Incubate linearizedplasmid + insert • for 30 min at 37 oC Transform cells E. coli enzymes do the rest
l Recombination system • Bacteriophage l • Double-stranded DNA bacteriophage • Lives as Dr Jekyll and Mr Hyde • 1) Lysogenic phase • Viral DNA integrated in host chromosome (“recombination”) • Not killing E. coli • Replicated together with E. coli genome • 2) Lytic phase • Phage replicates • Lyses E. coli cell after 45 min at 37 oC • Releasing ~100 progeny phages The l recombination system inserts phage DNA into E. coli genome
SLiCE is nice • High fidelity • Clean insertionby l phage recombination enzymes • Plasmid replicated by E. coli, not by PCR -> high fidelity 15 bp same in phage and E. coli DNA • Blunt ends are good: little accidental re-ligation of empty vectors
Summary • SLIC • Overhangs by exonuclease activity of T4 DNA polymerase • CPEC • Overlap extension by PCR • SLiCE • l recombination system • All need a linearized vector • By restriction enzyme • By overlap extension (PCR) • And an insert with matching ends • 20 complementary bases
x2 (or more) & & linear or circular multimers Anything that can happen will… + ligase & multimers of insert & either orientation