Download
1 / 65
650 likes | 763 Views
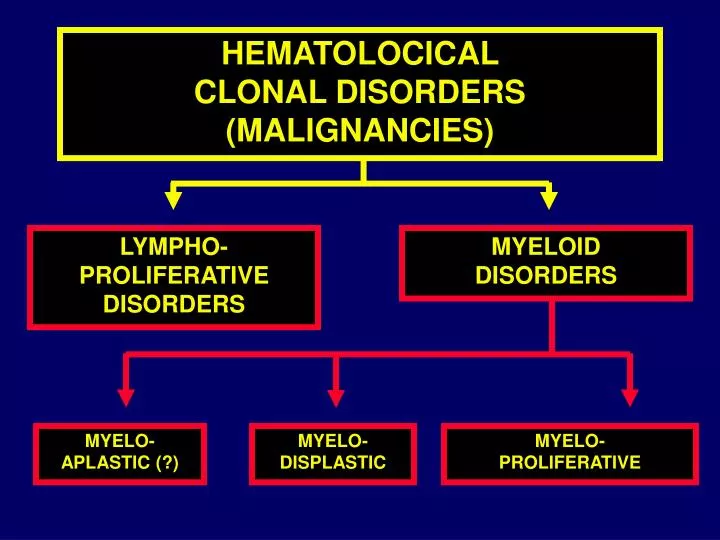
HEMATOLOCICAL CLONAL DISORDERS (MALIGNANCIES). LYMPHO- PROLIFERATIVE DISORDERS. MYELOID DISORDERS. MYELO- APLASTIC (?). MYELO- DISPLASTIC. MYELO- PROLIFERATIVE. MOLECULAR PATHS TO CANCER I. PROLIFERATION. APOPTOSIS. DIFFERENTIATION. Neoplasie della linea mieloide.

E N D
HEMATOLOCICAL CLONAL DISORDERS (MALIGNANCIES) LYMPHO- PROLIFERATIVE DISORDERS MYELOID DISORDERS MYELO- APLASTIC (?) MYELO- DISPLASTIC MYELO- PROLIFERATIVE
MOLECULAR PATHS TO CANCER I PROLIFERATION APOPTOSIS DIFFERENTIATION
Neoplasie della linea mieloide -Malattie mieloproliferative croniche - Sindromi mielodisplastiche Leucemia acuta mieloide Origine quasi sempre da cellula staminale pluripotente
NEOPLASIE MIELOIDI Mielopr. Cron. Differenziazione normale Sindr.Mielodispl. differenziazione Leuc. Ac. mieloide differenziazione
MYELO-PROLIFERATIVE DISORDERS • ACUTE • expansion of poorly differentiated elements • often with bone marrow functional failure • example: acute myeloid leukemiade novo • short survival if untreated • CHRONIC • expansion of well differentiated elements • from bone marrow failure to bone marrow hyperfunction • example; cronic myeloid leukemia, policitemia vera, essential thrombocytopenia • prolonged survival if left untreated • tend to loose differentiation with time
MYELOPROLYFERATIVE DISORDERS OFTEN INDUCED BY THE PRESENCE OF FUSION TRANSCRIPTS
FUSION TRANSCRIPTS: ARE A KIND OF DYSREGULATION THAT OCCURR PREFERENTIALY IN MYELOPROLIPHERATIVE DISORDERS IN MATURE LYMPHOID TUMORS, TRANSLOCATIONS OFTEN LEAD TO OVEREXPRESSION OF NORMAL GENES (except npm-alk)
CHRONIC MYELOPROLIFERATIVE DISORDERS CLONAL PROLIFERATION OF CELLS WITH GOOD DIFFERENTIATIVE POTENTIAL
MOLECULAR PATHS TO CANCER PROLIFERATION APOPTOSIS DIFFERENTIATION
CML IN CHRONIC PHASE PROLIFERATION (loss of physiological control) APOPTOSIS DIFFERENTIATION
Malattie mieloproliferative croniche Alterazione genetica Cellule staminali espansione Emopoiesi monoclonale, espansione dei progenitori, iperproduzione cellule mature, instabilità genetica Ulteriore alterazione genetica Leucemia acuta mieloide
Malattie mieloproliferative croniche Leucemia mieloide cronica: iperproduzione di granulociti +/- piastrine Mielofibrosi con metaplasia mieloide: iperproduzione di granulociti +/- piastrine (fase iniziale) Policitemia vera: iperproduzione di eritrociti, piastrine, granulociti Trombocitemia essenziale: iperproduzione di piastrine Tipiche dell’ età adulta: età mediana 55 – 65 anni
Myeloproliferative disorders CHRONIC MYELOGENOUSLEUKEMIA (CML) IDYIOPATIC MYELOFIBROSIS (IMMM) POLICITEMIAVERA (PV) ESSENTIAL THROBOCYTEMIA (TE) DISEASE SEVERITY GENETIC INSTABILITY
NATURAL HISTORY OF CMD CHRONIC MYELOGENOUSLEUKEMIA (CML) IDYIOPATIC MYELOFIBROSIS (FLORID PHASE) POLICITEMIAVERA ESSENTIAL THROBOCYTEMIA ACCELERATED PHASE SPENT (HYPERFIBROTIC)MPD MAJOR THROMBOTICEVENT BLASTIC (ACUTE) PHASE
High WBC High Hematocrit Large spleen Myeloid metaplasia Progression to AL Hypercellular BM Throbotic risk MPD CLINICAL FEATURES I Characteristic CML IMMM PV TE ++++ ++ ++ ± ± to - -- to + ++++ ± High PLTS +++ -- to ++ +++ ++++ +++ ++++ ++ ± ± to + +++ ++++ +++ ++++ -- +++ ++ +++ ++++ ++ ± ++++ ++ + ±
Prognosis (years) Knowledge of genetic basis MPD CLINICAL FEATURES I Characteristic CML IMMM PV TE 5-6* 10 15 20-25 ± ± ± ++++ ± ± ± ++++ Specific therapy *Pre-Glivec data
Trombocitemia essenziale Alterazioni cromosomiche molto rare. Espansione prevalente dei progenitori megacariocitari. Nel sangue piatrinosi > 600.000/ul, a volte molto elevata, > 1.500.000/ ul; raramente lieve leucocitosi con neutrofilia, quasi mai cellule immature circolanti. Midollo osseo normo-ipercellulare, con netto aumento dei megacariociti, spesso iperploidi Dopo circa 10 anni possibile evoluzione in mielofibrosi secondaria, raramente (1-5 % dei casi ) in LAM
Trombocitemia essenziale Sintomi: spesso paziente asintomatico (riscontro casuale); a volte sintomi di alterazioni del microcircolo: vertigini, acufeni, cefalea, parestesie alle estremità. Frequenti le complicanze vascolari trombotiche: infarto miocardio, ictus trombotico. Raramente diatesi emorragica. Diagnosi: escludere le piastrinosi secondarie (valutare sideremia, VES, cercare eventuali sintomi di neoplasie); escludere altre malattie mieloproliferative croniche: biopsia ossea, indagine molecolare per ricerca BCR/ABL.
Trombocitemia essenziale PROGNOSI E TERAPIA Sopravvivenza mediana circa 20 anni Causa di morte più frequente: incidenti vascolari Terapia anti-aggregante piastrinica se non vi è diatesi emorragica; citostatici (idrossiurea) se piastrine > 1.000.000 /ul o > 700.000 /ul e età > 60 anni o altri rischi vascolari; interferone alfa: idem se < 60 anni
Policitemia vera Alterazioni citogenetiche molto rare (20 q-) Ipersensibilità dei progenitori del clone mutato ad eritropoietina ed altre citochine (IL3, SCF). Aumento della massa eritrocitaria per iperproduzione: aumento progressivo Hb fino a valore > 20 g/dl ed Ht fino a > 60%; spesso anche piastrinosi moderata (400-800.000 /ul e lieve leucocitosi (10-20.000/ul) con neutrofilia. Non cellule immature circolanti. Raramente lieve splenomegalia. Cellularità midollare aumentata, fibrosi scarsa o assente. Nelle fasi avanzate (oltre 5-10 anni) tendenza a fibrosi midollare e riduzione della cellularità (evoluzione in mielofibrosi secondaria). 2 - 10% evoluzione in LAM.
Policitemia vera Sintomi e segni: conseguenti all’ iperviscosità ematica: vertigini, cefalea, dispnea da sforzo; rossore cutaneo, ipertensione; prurito dopo il bagno; alto rischio di incidenti vascolari: infarto miocardico, ictus trombotico, trombosi venosa profonda. In fasi avanzate possibili sintomi di citopenia (anemia)
Policitemia vera DIAGNOSI Escludere una eritrocitosi secondaria Criteri maggiori: massa eritrocitaria aumentata, O ht >64 normale saturazione di O2 dell’ Hb, splenomegalia Citogenetica alterata Esclusione di ogni altra causa Criteri minori: piastrinosi, leucocitosi, aumento fosfatasi alcalina leucocitaria, aumento vitamina B12 sierica. Richiesti 3 criteri maggiori o 2 maggiori + 2 minori. Attualmente importanti: dosaggio eritropoietina sierica (ridotta o ai limiti inferiori di norma), coltura in vitro progenitori eritroidi (crescita BFU-E senza aggiunta di eritropoietina)
Policitemia vera PROGNOSI E TERAPIA Sopravvivenza mediana 19 anni (più breve nei pazienti > 60 anni). Causa di morte più frequente: complicanze vascolari LAM più frequente nei pazienti che richiedono terapia citostatica. Trattamento principale è il salasso: ottenere Ht < 45% ; anti-aggreganti piastrinici se piastrinosi e/o precedenti trombosi. Chemioterapia citostatica (idrossiurea, pipobromano) indicata se piastrinosi + età >60 o altri rischi trombotici o insufficiente controllo dell’ eritrocitosi con salassi. Trattamento sperimentale: interferone alfa
Mielofibrosi con metaplasia mieloide Possibile anomalie citogenetiche non specifiche (+8, 20q-, 13q-). Espansione progenitori mieloidi e megacariocitari Emopoiesi extra- midollare (milza e fegato) Megacariocitopoiesi in parte inefficace Lisi intramidollare di megacariociti Liberazione fattori di crescita per fibroblasti ed endotelio (PDGF, VEGF) Progressiva fibrosi midollare e aumento emopoiesi extra-midollare: con il tempo citopenia ed epato/splenomegalia ingravescente
Mielofibrosi con metaplasia mieloide Fase iniziale: frequente leucocitosi, raramente piastrinosi; Hb normale o lievemente ridotta; splenomegalia lieve/moderata. Paziente asintomatico o lievi sintomi di splenomegalia; raramente sintomi infiammatori (febbre, artralgie, sudorazioni) Fase avanzata : anemia, piastrinopenia +/- leucopenia; splenomegalia imponente, sintomatica (dolori addominali, difficoltà all’ alimentazione). Sintomi di anemia e di splenomegalia, deperimento, complicanze infettive ed emorragiche. Evoluzione in LAM in circa il 20% dei casi
Mielofibrosi con metaplasia mieloide DIAGNOSI Sospetto per leucocitosi e/o splenomegalia. Esame dello striscio di sangue periferico: presenza di precursori granulocitari (mieloblasti- metamielociti, blasti < 5% ) ed eritroidi (eritroblasti); presenza di eritrociti a lacrima (dacriociti) Aspirato midollare:quasi sempre “punctio sicca” Biopsia ossea: cellularità e fibrosi di grado variabile secondo lo stadio, iperplasia megacariocitaria con displasia. Biologia molecolare: escludere presenza di BCR/ABL
Mielofibrosi con metaplasia mieloide PROGNOSI Sopravvivenza mediana 4 - 5 anni, ma molto variabile: 2-3 anni se anemia (Hb < 10 g/dl) o sintomi infiammatori o GB > 30.000 /ul o blasti circolanti >3% > 10 anni se nessuno dei fattori prognostici negativi TERAPIA Fase iniziale: idrossiurea se leucocitosi, piastrinosi o splenomegalia. Fase avanzata: trasfusioni, androgeni, eventuale splenectomia o radioterapia splenica. Terapie sperimentali: trapianto di midollo, eritropoietina ricombinante
CML: A MODEL FOR CANCER PATHOGENESIS The first neoplasms treatable with a smart drug
LEUCEMIA MIELOIDE CRONICA (LMC) PATOGENESI Nel 90 % dei casi presente traslocazione cromosomica t (9;22): cromosoma Filadelfia (Ph1), con scambio tra i geni BCR e ABL. Sul cromosoma 22 formazione di gene ibrido BCR/ABL, che codifica per proteina di fusione BCR/ABL con spiccata attività tirosino-chinasica.
LEUCEMIA MIELOIDE CRONICA PATOGENESI In 5% dei casi fusione genica BCR/ABL senza riscontro citogenetico del cromosoma Ph1 (casi Ph1- ma BCR/ABL+): stessa malattia, prognosi e risposta terapeutica. Solo 5% dei casi è BCR/ABL negativo: LMC atipiche, molto rare e a prognosi peggiore.
LEUCEMIA MIELOIDE CRONICA PATOGENESI La tirosino-chinasi BCR/ABL fosforila diversi substrati, riproducendo gli effetti di una stimolazione continua con fattori di crescita . vantaggio proliferativo della cellula staminale mutata e della sua progenie e instabilità genetica che predispone a ulteriori mutazioni. L’ acquisizione di ulteriori mutazioni porta all’ evoluzione a leucemia acuta.
PATOGENESI LMC 9 q 22 q bcr abl 9 q 22 q bcr/abl abl/bcr ininfluente\ Tirosino-chinasi
PATOGENESI LMC Chinasi BCR/ABL fosforilazione Attivazione ras stimolo proliferativo Effetto anti- apoptotico Ridotta adesività a stroma Instabilità genetica Perdita sensibilità a inibitori proliferazione
CP-CML CELLS • REQUIRE LESS SURVIVAL/PROLFERATIVE SIGNALS BY STROMAL CELLS • DO NOT REQUIRE ADHESIVE MOLECULES • IMPAIR DIFFERENTIATION AND SURVIVAL OF THEIR NORMAL COUNTERPARTS • LESS SENSITIVE TO INHIBITORY/APOPTOTIC STIMULI
The abl-gene • homologous to v-abl • IN G1 it is bound by Rb1 • Knock-out mice are leukopenic • when overexpressed --> cell cycle arrest ABL protein ATP bind SH3 SH2 SH1 NLS DNA BINDING ACTIN BINDING NH2 COOH Protein interaction Tyr Kinase NLS: Nuclear Localisation Signal
The BCR-gene • Ubiquitous expression • cytoplasmic, but pericromosomic during mitosis • Knock-out have nearly normal hematopoiesis BCR protein DD Ser/Trh kinase DBL/GAP PH COOH NH2 177Y Sh2 binding Dimerization domain
Characteristics of bcr-abl • Only cytoplasmic • permanently activated • several targets • it mimics some TK receptors, inducing constitutive activation of several pathways • bypass of regulatory mechanisms
Apoptosis inhibition Proliferative stimulus
Malattie mieloproliferative croniche Alterazione genetica Cellule staminali espansione Emopoiesi monoclonale, espansione dei progenitori, iperproduzione cellule mature, instabilità genetica Ulteriore alterazione genetica Leucemia acuta mieloide
A TRIPHASIC DISEASE ACCELERATED PHASE BLASTIC PHASE CHRONIC PHASE
LEUCEMIA MIELOIDE CRONICA: STORIA NATURALE Fase cronica: 4 anni (1 - >10). -Leucocitosi: 20 - 100.000 / l nei casi a diagnosi occasionale, 100 - > 400.000 nei casi sintomatici, con neutrofilia; -frequente basofilia e costante presenza di precursori granulocitari (promielociti – metamielociti, blasti 1 –5% nei casi con iperleucocitosi). Frequente piastrinosi moderata (400.000 – 1.000.000/ l), raramente iperpiastrinosi (> 1.000.000/ l) e leucocitosi minima.
LEUCEMIA MIELOIDE CRONICA: STORIA NATURALE Fase cronica - Eritrociti nella norma o lieve anemia. Splenomegalia frequente, che correla con l’ entità della leucocitosi. Midollo osseo ipercellulare, con spiccata iperplasia granulocitopoietica, normale maturazione (blasti < 5%), aumento dei megacariociti; fibrosi assente o scarsa. - Sintomi assenti o aspecifici: astenia, dimagrimento, sudorazioni, tensione o dolori addominali da splenomegalia, nei casi con iperleucocitosi.
LEUCEMIA MIELOIDE CRONICA: STORIA NATURALE Fase accelerata Durata 6 – 12 mesi; può anche mancare nei casi a trasformazione acuta. Possibile comparsa di ulteriori alterazioni del cariotipo. Iniziali segni di alterata maturazione emopoietica: aumento cellule immature nel sangue e nel midollo, spesso anemia, piastrinopenia o piastrinosi, incremento della basofilia. Frequente incremento della splenomegalia, indipendente dalla leucocitosi. -Frequente accentuazioni di sintomi infiammatori: astenia, sudorazioni, febbre, dolori ossei.
LEUCEMIA MIELOIDE CRONICA: STORIA NATURALE Fase blastica -Sopravvivenza di pochi mesi. -Frequenti anomalie aggiuntive del cariotipo (due cromosomi Ph1, +8, inversione del 17). -Quadro ematologico di leucemia acuta (mieloide nel 70%, linfoide nel 30% dei casi): leucocitosi con blasti midollari o periferici > 20%, anemia, piastrinopenia, granulocitopenia. Frequente epato-splenomegalia, a volte adenopatie. - Frequenti sintomi di anemia, dolori addominali per splenomegalia, febbre, dimagrimento, infezioni e diatesi emorragica. Morte di solito per infezioni o emorragia.
CML IN BLASTIC PHASE PROLIFERATION (loss of physiological control) APOPTOSIS DIFFERENTIATION
CML EVOLUTION TELOMERE SHORTENING AND TELOMERASE ACTIVATION P53 INCTIVATION EPIGENETIC SILENCING ADDITIONAL TRANSLOCATIONS