Download

1 / 50
560 likes | 1.05k Views
Growth Disorders. Most children evaluated for short stature are normal stature variants . The slow growth patterns of these children reflect familial short stature and/or constitutional delay of growth and puberty. Familial short stature. Tend to be small at birth

E N D
Most children evaluated for short stature are normal stature variants. • The slow growth patterns of these children reflect familial short stature and/or constitutional delay of growth and puberty
Familial short stature • Tend to be small at birth • They grow approximately parallel to the normal curve but below the 3rd percentile from infancy • Pubertal development and the acceleration of growth occur normally and they ultimately achieve a short adult height(Father <163 cm,Mother <150 cm)
They have no clinical or laboratory evidence of endocrine or systemic disease • Their annual growth rates are within normal limits • The bone age is appropriate for his or her chronological age
The diagnosis of familial short stature has been perpetuated in families in which impaired growth was eventually proved to be consequence of a defined genetic disorder.
Constitutional delay in growth and puberty(CDGP) • They mature at a slower than normal rate • Height and bone age are usually delayed by 2-4 years • Onset of pubertal maturation is understandably delayed appropriate to the child’s bone age • Often there is a history of delay in growth and pubertal development of either parent or another relative • Final adult stature, which may not be reached until the age of 20 or more years, is appropriate for parental target height range
They are usually of normal size at birth • They grow normally for several months or years but then deviate from the “normal” growth centiles, particularly in the pre/peri-pubertal years • They grow at a normal rate for their bone age • There is a later than average adolescent “growth spurt” • Treatment: controversial • Synthetic steroids designed to have an increased anabolic-androgenic ratio:oxandrolone(oral)(hepatotoxic-hepatic tumors!) • No suitable equivalent for the girl
A child’s current centile must be evaluated in terms of: • Genetic background • Gestational or past medical history • Environment • Physical findings • Growth pattern since birth • Current growth rate(height velocity)
Causes of short stature 1- Familial or congenital conditions • Skeletal dysplasias • Chromosomal abnormalities • Forms of intrauterine growth retardation which may result in show or abnormal growth throughout later life 2- Chronic systemic disorders: • (Due to) insufficient intake of energy and/or protein-nutritional insufficiency, malabsorption syndromes and chronic inflammatory bowel disease) • Insufficient oxygenation of tissues • Electrolyte imbalance(chronic renal failure)
3-Endocrine abnormalities • CNS or hypothalamic abnormalities which affect regulation of the anterior pituitary • Anterior pituitary disorders, either developmental or acquired which can affect synthesis or secretion of growth hormone or the trophic hormones(TSH,LH,FSH,ACTH) • Primary dysfunction of the target organs e.g. Thyroid,adrenal or gonadal disease
Peripheral target tissue abnormalities - a diminished or absent hormone receptor or tissue enzyme defect Laron type “dwarfism” – failure of IGF1 generation in response to GH (A GH receptor defect) - Disorders related to the interaction of the IGF’s or other related peripheral growth factors with chondrocytes and mucopolysaccharides in epiphyseal growth cartilage Pygmies:genetic defect in IGF-1 responsiveness associated with diminished IGF-1 binding
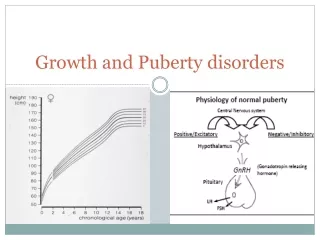
Normal human growth hormone secretion • In the first days of life, very high levels of GH occur. After 2 weeks of age, lower mean levels are found. • In pubertal children, the basal plasma GH concentration is not significantly different from that reported for adults but more peaks of GH may occur during the day with greater amplitude of GH peaks during the night • The most consistent period of GH secretion for both children and adults occurs within one hour or so after the onset of sleep.The initial surge of GH secretion often correlates with the onset of stage 3 or 4 (slow wave) sleep
Control of human growth hormone secretion • Positive releasing factor GHRH-Ghrelin • Inhibitory factor:GHRIH • Naturally occurring events that trigger GH secretion in man are exercise, physical and emotional stresses and high protein intake
Normal regulation of GH • The neurone system regulating GHRH and GHRH release receives a variety of normal impulses.Impulses arising in the hippocampus are stimulatory whereas those arising in the amygdaloid nuclei can be either stimulatory or inhibitory. The inhibitory inputs are presumed to activate GHRIH release via the anterior somatostinergic pathways,the stimulatory pathway is by way of the ventromedial(VM) nucleus of the hypotalamus
The GH regulatory neurone system also recieves impulses from each of the ascending monoaminergic neuronal systems : dopaminergic, noradrenergic and serotonergic - L-Dopa(converted to dopamine) in the brain leads to a release of GH - Sleep induced GH release is predominantly mediated by serotonergic fibres. - Hypoglycemia induced GH release is mediated by noradrenergic pathways.
GH provocation tests • Exercise • Sleep • Arginine infusion • Glucagon • Clonidine (alfa adrenergic receptor stimulant) • ITT • L-Dopa • Priming(sex hormones)??
Physiologic effects of human growth hormone • Anabolic-promotes protein synthesis in muscle cells • Catabolic effect- on fat and carbonhydrate metabolism (short term hypoglycemic effect) inducing lipolysis in adipoctytes • Long term effect of high GH levels:plasma glucose concentration increases(increased glucose production and reduced glucose utilization)
Evalution of height measurements 1- Growth charts 2- Parental heights- target height 3- Height SDS 4- Height velocity 5- Predicted ht SDS:X-x* SD x*:mean height for age and gender
Etiology and pathogenesis of growth failure • Chromosomal assessment Turner Sydrome • FSS • IUGR “Catch up” • CDGP • Skeletal dysplasia osteochondrodysplasias(achondroplasia, hypochondroplasia)
Chronic systemic disorders- asthma • Nutritional insufficiency • Chronic gastrointestinal diseases Coeliac disease Crohn’s disease Ulcerative colitis • Chronic renal disease • Cardiac disease • DM
Endocrine abnormalities • Primary hypothyrodism • Gonadal dysgenesis • GHD • Glucocorticoid excess • Growth failure to pyschosocial deprivation
Etiology of GHD • Congenital Acquired • İsolated MPHD
Incidence 1/4000-10000 live births • (50-60%) idiopathic • Hereditary (aut.recessive or dominant)
Developmental defects • Pituitary aplasia-hypoplasia • Midline abnormalities - Severe holoprosencephaly septooptic dysplasia (SOD):Hypoplasia of the optic nerves dysgenesis or agenesis of the infundibulum and septum pellucidum +/- pituitary hormone deficiency -Cleft lip palate -Single upper central incisor syndrome
Fizik Muayene-1: • TY: 11 yaş 11 ay iken • Boy: 131 cm • Boy SDS: - 3.4 • VA: 28 kg • BMI: 16,32 kg/m² • KY: 11-12 YAŞ
BH gene defects • All patients with classical GH gene deletions have complete GHD(IGHD IA, IB,2,3(+hypogammaglobulinemic))
GH gene regulation: • Pit1 (pituitary transcription factor):stimulatory effect on GH gene transcription • Pit 1 mutations - Severe deficiencies in GH PRI - Often develop secondary hypothyrodism • Prop 1 (Prophet of pit 1) mutations combined pituitary hormone deficiency(secondary hypogonadism)
Clinical features of GHD • Growth failure • Normal birth weight and birth length • Skeletal proportions normal for age • Somewhat overweight for height(increased subcutaneous fat) • Head circumference normal • Disparity between the size of face and the calvarium: “doll like” “cherubic” facies most pronounced in panhypopituitarism (crowding of the facial features to the centre of the face suggesting maxillary hypoplasia)- frontal bossing, saddle nose • Skeletal maturity delayed • High pitched voice • Males: penis,scrotum small, hypoplastic testis • Hypoglycemia
Radiological assessment: Skull x rays,CT, MRI • IGF 1 measurement, physiological tests of GH secretion