Download

1 / 29
330 likes | 750 Views
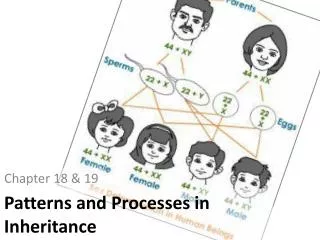
LP7 - Inheritance / Heredity and Genetic Diseases. Inheritance patterns: Monogenic (Mendelian) Inheritance Polygenic and Multifactorial Inheritance Mitochondrial Inheritance. Inheritance patterns.

E N D
LP7 - Inheritance / Heredity and Genetic Diseases Inheritance patterns: Monogenic (Mendelian) Inheritance Polygenic and Multifactorial Inheritance Mitochondrial Inheritance
Inheritance patterns Inheritance patterns trace the transmission of genetically encoded traits, conditions or diseases to offspring. There are several modes of inheritance: • Single Gene or Mendelian • Polygenic and Multifactorial • Mitochondrial
Single Gene Inheritance • Genetic conditions caused by a mutation in a single gene follow predictable patterns of inheritance within families. Single gene inheritance is also referred to as Mendelian inheritance as they follow transmission patterns he observed in his research on peas. • There are four types of Mendelian inheritance patterns: • Autosomal: the gene responsible for the phenotype is located on one of the 22 pairs of autosomes (non-sex determining chromosomes). • X-linked: the gene that encodes for the trait is located on the X chromosome. • Dominant: conditions that are manifest in heterozygotes (individuals with just one copy of the mutant allele). • Recessive: conditions are only manifest in individuals who have two copies of the mutant allele (are homozygous). • Y-linked (holandric): the gene that encodes for the trait is located on the Y chromosome
Autosomal dominant (AD) • Dominant conditions are expressed in individuals who have just one copy of the mutant allele. • The pedigree on the right illustrates the transmission of an autosomal dominant trait. • Affected males and females have an equal probability of passing on the trait to offspring. • Affected individual’s have one normal copy of the gene and one mutant copy of the gene, thus each offspring has a 50% chance on inheriting the mutant allele. • As shown in this pedigree, approximately half of the children of affected parents inherit the condition and half do not. • Huntington Disease · Myotonic muscular dystrophy • Acondroplasia (short-limbed dwarfism) • Polycystic kidney disease (PKDU) · Brachydactyly · Polydactily · Syndactyly · Adactyly • Osteogenesis imperfecta · Gout · Familial hypercholesterolemia · Hypercalcemia · Marfan syndrome · Familial polycystitis · Neurofibromatosis
Autosomal dominant (AD) • Huntington Disease • Myotonic muscular dystrophy • Achondroplasia (short-limbed dwarfism) • Polycystic kidney disease (ADPKD) • Brachydactyly • Polydactily • Syndactyly • Adactyly • Osteogenesis imperfecta • Gout • Familial hypercholesterolemia • Hypercalcemia (familial) • Marfan syndrome • Familial Polycystic ovary syndrome (PCOS) • Neurofibromatosis
Huntington Disease • Huntington's disease (HD) is a neurodegenerative genetic disorder that affects muscle coordination and leads to cognitive decline and psychiatric problems. It typically becomes noticeable in mid-adult life. HD is the most common genetic cause of abnormal involuntary writhing movements called chorea, which is why the disease used to be called Huntington's chorea. • The Huntingtin gene (HTT=HD=IT15) on 4p16.3 provides the genetic information for a protein that is also called "huntingtin". Expansion of a CAG triplet repeat stretch within the Huntingtin gene results in a different (mutant) form of the protein, which gradually damages cells in the brain, through mechanisms that are not fully understood. The genetic basis of HD was discovered in 1993 by an international collaborative effort spearheaded by the Hereditary Disease Foundation.
Huntington Disease • Increases in the number of repeats (and hence earlier age of onset and severity of disease) in successive generations is known as genetic anticipation. Instability is greater in spermatogenesis than oogenesis; • Individuals with more than sixty repeats often develop the disease before age 20, while those with fewer than 40 repeats may not ever develop noticeable symptoms; • Life expectancy in HD is generally around 20 years following the onset of visible symptoms; • Most life-threatening complications result from muscle coordination and, to a lesser extent, behavioral changes induced by declining cognitive function. • The largest risk is pneumonia, which causes death in one third of those with HD. As the ability to synchronize movements deteriorates, difficulty clearing the lungs and an increased risk of aspirating food or drink both increase the risk of contracting pneumonia. The second greatest risk is heart disease, which causes almost a quarter of fatalities of those with HD.[
Huntington Disease • Recommended (highly) to see what Huntington is all about • An excellent French documentary (subtitled in English) about a family carrying such a genetic “burden”, including aspects of their life and expectancies • As a reminder, the disease has a complete penetrance (100%) make the disease, usually after 35-40 years of age, and transmit it to their progenitors • http://www.youtube.com/watch?v=0qOdGvoOXI0(it takes 1 hour and a half)
Other AD conditions • Myotonic muscular dystrophy (dystrophia myotonica, myotonia atrophica) is a chronic, slowly progressing, highly variable, inherited multisystemic disease. It is characterized by wasting of the muscles (muscular dystrophy), cataracts, heart conduction defects, endocrine changes, and myotonia. • Achondroplasia is a common cause of dwarfism. It occurs as a sporadic mutation in approximately 75% of cases (associated with advanced paternal age) or may be inherited as an autosomal dominant genetic disorder. People with achondroplasia have short stature, with an average adult height of 131 centimeters for males and 123 centimeters for females. Achondroplastic adults are known to be as short as 62.8 cm (24.7 inches) • Polycystic kidney disease (PKD or PCKD, also known as polycystic kidney syndrome) is a cystic genetic disorder of the kidneys.There are two types of PKD: autosomal dominant polycystic kidney disease (ADPKD) and the less-common autosomal recessive polycystic kidney disease (ARPKD). Polycystic kidney disease is one of the most common life-threatening genetic diseases, affecting an estimated 12.5 million people worldwide.
Other AD conditions • Brachydactyly (short fingers/toes) • Polydactily (extra fingers/toes) • Syndactyly (two or more digits are fused together) • Adactyly (congenital absence of fingers and/or toes) • Osteogenesis imperfecta (OI and sometimes known as brittle bone disease, or "Lobstein syndrome") is a congenital bone disorder. People with OI are born with defective connective tissue, or without the ability to make it, usually because of a deficiency of Type-I collagen. As a genetic disorder, OI has historically been viewed as an autosomal dominant disorder of type I collagen. In the past several years, there has been the identification of autosomal recessive forms.Most people with OI receive it from a parent but in 35% of cases it is an individual (de novo or "sporadic") mutation. There are eight different types of OI, Type I being the most common, though the symptoms vary from person to person.
Other AD conditions • Gout (also known as podagra when it involves the big toe). is a medical condition usually characterized by recurrent attacks of acute inflammatory arthritis—a red, tender, hot, swollen joint. The metatarsal-phalangeal joint at the base of the big toe is the most commonly affected (approximately 50% of cases). However, it may also present as tophi, kidney stones, or urate nephropathy. It is caused by elevated levels of uric acid in the blood. The uric acid crystallizes, and the crystals deposit in joints, tendons, and surrounding tissues. The occurrence of gout is partly genetic, contributing to about 60% of variability in uric acid level.[ • Familial hypercholesterolemia (abbreviated FH) is a genetic disorder characterized by high cholesterol levels, specifically very high levels of low-density lipoprotein (LDL, "bad cholesterol"), in the blood and early cardiovascular disease. Many patients have mutations in the LDLR gene that encodes the LDL receptor protein, which normally removes LDL from the circulation, or apolipoprotein B (ApoB), which is the part of LDL that binds with the receptor; mutations in other genes are rare. Patients who have one abnormal copy (are heterozygous) of the LDLR gene may have premature cardiovascular disease at the age of 30 to 40. Having two abnormal copies (being homozygous) may cause severe cardiovascular disease in childhood. Heterozygous FH is a common genetic disorder, inherited in an autosomal dominant pattern, occurring in 1:500 people in most countries; homozygous FH is much rarer, occurring in 1 in a million births.
Other AD conditions • Hypercalcemia - Familial hypocalciuric hypercalcemia is a condition that can cause hypercalcemia, a serum calcium level typically above 10.2 mg/dL. It is also known as familial benign hypocalciuric hypercalcemia (FBHH) where there is usually a family history of hypercalcemia which is mild, a urine calcium to creatinine ratio <0.01, and urine calcium <200 mg/day. • Familial Polycystic ovary syndrome (PCOS) is one of the most common female endocrine disorders. PCOS is a complex, heterogeneous disorder of uncertain etiology, but there is strong evidence that it can to a large degree be classified as a genetic disease. PCOS produces symptoms in approximately 5% to 10% of women of reproductive age (12–45 years old). It is thought to be one of the leading causes of female subfertilityand the most frequent endocrine problem in women of reproductive age. The genetic component appears to be inherited in an autosomal dominant fashion with high genetic penetrance but variable expressivity in females; this means that each child has a 50% chance of inheriting the predisposing genetic variant(s) from a parent, and if a daughter receives the variant(s), then the daughter will have the disease to some extent.[
Other AD conditions • Marfan syndrome (also called Marfan's syndrome) is a genetic disorder of the connective tissue. People with Marfan tend to be unusually tall, with long limbs and long, thin fingers. The syndrome is inherited as a dominant trait, carried by the gene FBN1, which encodes the connective protein fibrillin-1. People have a pair of FBN1 genes. Because it is dominant, people who have inherited one affected FBN1 gene from either parent will have Marfan syndrome. Marfan syndrome has a range of expressions, from mild to severe. The most serious complications are defects of the heart valves and aorta. It may also affect the lungs, the eyes, the dural sac surrounding the spinal cord, the skeleton and the hard palate.
Other AD conditions • Neurofibromatosis (commonly abbreviated NF; neurofibromatosis type 1 is also known as von Recklinghausen disease) is a genetically-inherited disorder in which the nerve tissue grows tumors (neurofibromas) that may be benign and may cause serious damage by compressing nerves and other tissues. Neurofibromatosis is an autosomal dominant disorder, which means only one copy of the affected gene is needed for the disorder to develop. Therefore, if only one parent has neurofibromatosis, his or her children have a 50% chance of developing the condition as well. The severity in affected individuals can vary; this may be due to variable expressivity. Approximately half of cases are due to de novo mutations and no other affected family members are seen. It affects males and females equally.
Autosomal Recessive (AR) • Recessive conditions are clinically manifest only when an individual has two copies of the mutant allele. • When just one copy of the mutant allele is present, an individual is a carrier of the mutation, but does not develop the condition. • Females and males are affected equally by traits transmitted by autosomal recessive inheritance. • When two carriers mate, each child has a 25% chance of being homozygous wild-type (unaffected); a 25% chance of being homozygous mutant (affected); or a 50% chance of being heterozygous (unaffected carrier). Note: Affected individuals are indicated by solid black symbols and unaffected carriers are indicated by the half black symbols • Cystic fibrosis · Phenylketonuria (PKU) · Albinism · Galactosemia · Xeroderma pigmentosum · Fanconi anemia · Bloom syndrome • Tay-Sachs • Hemochromatosis
Autosomal Recessive (AR) • Cystic fibrosis • Phenylketonuria (PKU) • Albinism • Galactosemia • Xeroderma pigmentosum • Fanconi anemia • Bloom syndrome • Tay-Sachs • Hemochromatosis
X-linked Dominant • Because the gene is located on the X chromosome, there is no transmission from father to son, but there can be transmission from father to daughter (all daughters of an affected male will be affected since the father has only one X chromosome to transmit). • Children of an affected woman have a 50% chance of inheriting the X chromosome with the mutant allele. • X-linked dominant disorders are clinically manifest when only one copy of the mutant allele is present. • Some forms of Retinitis Pigmentosa • Chondrodysplasia Punctata • Hypophosphatemic rickets, also called X-linked hypophosphatemia (XLH), hypophosphatemic vitamin D-resistant rickets (HPDR) · Amelogenesis imperfecta
X-linked Dominant • Some forms of Retinitis Pigmentosa • Chondrodysplasia Punctata • Hypophosphatemic rickets = X-linked hypophosphatemia (XLH) =Hypophosphatemic vitamin D-resistant rickets (HPDR) • Amelogenesis imperfecta
X-linked Recessive • X-linked recessive traits are not clinically manifest when there is a normal copy of the gene. • All X-linked recessive traits are fully evident in males because they only have one copy of the X chromosome, thus do not have a normal copy of the gene to compensate for the mutant copy. • For that same reason, women are rarely affected by X-linked recessive diseases, however they are affected when they have two copies of the mutant allele. • Because the gene is on the X chromosome there is no father to son transmission, but there is father to daughter and mother to daughter and son transmission. • If a man is affected with an X-linked recessive condition, all his daughter will inherit one copy of the mutant allele from him. • Duchenne muscular dystrophy (DMD) • Hemophilia A • X-linked severe combined immune disorder (SCID) • Some forms of congenital deafness
Y-linked (holandric traits) • Hypertrichosis of the ears
Polygenic and Multifactorial Inheritance (1) • Most diseases have multifactorial inheritance patterns. • As the name implies, multifactorial conditions are not caused by a single gene, but rather are a result of interplay between genetic factors and environmental factors. • Diseases with multifactorial inheritance are not genetically determined, but rather a genetic mutation may predispose an individual to a disease. Other genetic and environmental factors contribute to whether or not the disease develops. • Numerous genetic alterations may predispose individuals to the same disease (genetic heterogeneity). • For instance coronary heart disease risk factors include high blood pressure, diabetes, and hyperlipidemia. All of those risk factors have their own genetic and environmental components. Thus multifactorial inheritance is far more complex than Mendelian inheritance and is more difficult to trace through pedigrees. DISEASES: • Alzheimers disease • Heart disease • Some cancers • Neural tube defects • Schizophrenia • Insulin-dependent Diabetes mellitus CHARACTERS: · Height, weight • Intelligence · Skin, eyes and hair color · Dermatoglyphics · Blood pressure
Polygenic and Multifactorial Inheritance (2) • A typical pedigree from a family with a mutation in the BRCA1 gene. • Fathers can be carriers and pass the mutation onto offspring. • Not all people who inherit the mutation develop the disease, thus patterns of transmission are not always obvious.
Polygenic and Multifactorial Inheritance (3) • Alzheimers disease • Heart disease • Some cancers • Neural tube defects • Schizophrenia • Insulin-dependent diabetes mellitus • Height, weight • Intelligence • Skin, eyes and hair color • Dermatoglyphics • Blood pressure
Mitochondrial Inheritance (1) • Mitochondria are organelles found in the cytoplasm of cells. • Mitochondria are only inherited from the mother’s egg, thus only females can transmit the trait to offspring, however they pass it on to all of their offspring. • The primary function of mitochondria is conversion of molecule into usable energy. • Thus many diseases transmitted by mitochondrial inheritance affect multiple organs with high-energy use such as the heart, blood, skeletal muscle, liver, and kidneys, becoming a complex texture of diseases, usually lethal in early childhood. • The difficulty arises when no mtDNA defect can be found or when the clinical abnormalities are complex and not easily matched to those of more common mitochondrial disorders.
Mitochondrial Inheritance (2) • Mitochondria are unique in that they have multiple copies of a circular chromosome = mtDNA Each human cell contains thousands of copies of mtDNA. At birth these are usually all identical (homoplasmy). By contrast, individuals with mitochondrial disorders resulting from mtDNA mutations may harbor a mixture of mutant and wild-type mtDNA within each cell (heteroplasmy) The percentage level of mutant mtDNA may vary among individuals within the same family, and also among organs and tissues within the same individual. This is one explanation for the varied clinical phenotype seen in individuals with pathogenic mtDNA disorders.