Download

1 / 128
1.28k likes | 1.6k Views
第三章 外周神经系统药物 Peripheral Nervous System Drugs. 内容包括:. 第一节 拟胆碱药 第二节 抗胆碱药 第三节 拟肾上腺素药 第四节 组织胺 H1 受体拮抗剂 第五节 局部麻醉药. 毒蕈碱型受体( M 受体 : M1 , M2 , M3 , M4 , M5 ). 胆碱受体. 烟碱型受体( N 受体 :N1,N2 ). 第一节 拟胆碱药 Cholinergic Drugs. M 受体,心脏活动抑制( M2 ),平滑肌收缩( M3 ) ,腺体分泌增加( M1 、 M3 ) ,瞳孔缩小( M3 )等。

E N D
第三章 外周神经系统药物 Peripheral Nervous System Drugs 内容包括: 第一节 拟胆碱药 第二节 抗胆碱药 第三节 拟肾上腺素药 第四节 组织胺H1受体拮抗剂 第五节 局部麻醉药
毒蕈碱型受体(M受体: M1,M2,M3, M4,M5) 胆碱受体 烟碱型受体(N受体:N1,N2) 第一节 拟胆碱药Cholinergic Drugs
M受体,心脏活动抑制(M2),平滑肌收缩(M3)M受体,心脏活动抑制(M2),平滑肌收缩(M3) ,腺体分泌增加(M1、M3),瞳孔缩小(M3)等。 老年痴呆(M1) N1位于神经节突触后膜,可引起自主神经节的 节后神经元兴奋,肾上腺素释放增加。 N2受体位于骨骼肌终板膜,可引起运动终板电 位,导致骨骼肌收缩。
拟胆碱药指一类作用与乙酰胆碱相似的药物 • 直接拟胆碱药:作用并兴奋Ach-受体 • (M 受体/N 受体) • 间接拟胆碱药:抑制Ach-E,使Ach水解受到 • 抑制,增加内源性Ach在突触间隙中的量
人工合成的 可逆的 天然生物碱 不可逆的 分类 • M受体激动剂胆碱酯酶抑制剂
一、M受体激动剂muscarinic receptor agonists • M样作用:引起心肌收缩力减弱,心率减慢;消化道、呼吸道及其他脏器平滑肌收缩;动脉血管平滑肌松弛,血管舒张,但大剂量又可使静脉血管收缩;腺体分泌增加。 • M受体激动剂属于直接作用于胆碱受体的拟胆碱药。 • M受体激动剂主要用于手术后腹气涨、尿潴留;降低眼内压,治疗青光眼;大部分胆碱受体激动剂还具有吗啡样镇痛作用,可用于止痛。
一)人工合成的乙酰胆碱类似物 • ACh对所有胆碱能受体部位无选择性,导致产生副作用。 • ACh为季铵结构,不易透过生物膜,因此生物利用度极低。 • ACh化学稳定性差,在水溶液、胃肠道和血液中均易被水解或胆碱酯酶催化水解,失去活性。 乙酰胆碱(ACh)
方法要点: 亚乙基上增加一个甲基取代,水解↓。 乙酰基→氨乙酰基,酯基稳定性↑。
胆碱酯类M受体激动剂的构效关系 五原子规则 氨甲酰基取代使酯键稳定 S-异构体的活性大大高于R-异构体
毛果芸香碱 , Pilocarpine ,叔胺类化合物。在体内以质子化的季铵正离子为活性形式。两个手性碳,3S-cis。 用途:具缩瞳、降低眼内压作用(M1,M3)。 用其硝酸盐或盐酸盐制成滴眼液治疗原发性青光眼
毛果芸香碱的稳定性 水解和3-差向异构,失活
毛果芸香碱的衍生药物 方法一:制成前药,生物利用度↑,化学稳定性↑ 方法二:氨甲酸酯类似物 ,不易水解失活,长效
三)选择性M受体亚型激动剂 • 西维美林 Cevimeline (M1/M3 ) 2000年上市,口腔干燥症 • 呫诺美林 Xanomeline (M1 ),槟榔碱衍生物 阿尔茨海默病
二、乙酰胆碱酯酶抑制剂Acetylcholinesterase Inhibitors • 乙酰胆碱酯酶的结构及其水解乙酰胆碱的机理 • 可逆性乙酰胆碱酯酶抑制剂 • 不可(难)逆性乙酰胆碱酯酶抑制剂
ACh-AChE 可逆复合物 乙酰化酶 广义碱催化乙 酰化酶的水解 游离酶 乙酰胆碱酯酶催化乙酰胆碱水解机制 失活 复活
乙酰胆碱酯酶催化乙酰胆碱水解机制 乙酸酯 几十毫秒
可逆性乙酰胆碱酯酶抑制剂 • 生物碱类:毒扁豆碱 • 季铵类:溴新斯的明 • 叔胺类:盐酸多奈哌齐 • 其他类
溴新斯的明Neostigmine Bromide 溴化-N,N,N-三甲基-3-[(二甲氨基)甲酰氧基]苯铵 N,N,N-三甲基-3-[(二甲氨基)甲酰氧基]苯铵 用于重症肌无力和术后腹气胀及尿潴留。
溴新斯的明的发现 第一个用于临床的抗胆碱酯酶药,选择性↓,毒性↑,中枢副作用。
结构特点 中枢作用↓,稳定性↑,口服,注射。
代谢 主要代谢物是酯水解产物溴化3-羟基苯基三甲铵,具有与Neostigmine相似但较弱的活性。
与胆碱酯酶的相互作用过程 几分钟 氨基甲酸酯
同型药物 溴新斯的明 Neostigmine Bromide 溴吡斯的明 Pyridostigmine Bromide 苄吡溴铵 Benzpyrinium Bromide 地美溴铵 Demecarium Bromide
非经典的抗胆碱酯酶药--抗AD药 他克林 Tacrine ,1993 多奈哌齐 Donepezil ,1997 卡巴拉汀 ,2000 Rivastigmine
非经典的抗胆碱酯酶药--抗AD药 加兰他敏 Galantamine 石杉碱甲 Huperzine A 美曲膦酯 Metrifonate 敌敌畏 Dichlorvos,DDVP 不可逆
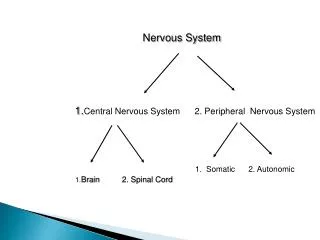
第二节 抗胆碱药Anticholinergic Drugs • M受体拮抗剂 • N受体拮抗剂
一、 M受体拮抗剂 muscarinic receptor antagonists • 作用:抑制腺体(唾液腺、汗腺、胃液)分泌,散大瞳孔,加速心律,松弛支气管和胃肠道平滑肌等作用。临床用于治疗消化性溃疡、散瞳、平滑肌痉挛导致的内脏绞痛等。 • 分类: 1)天然茄科生物碱类及其半合成类似物 2)合成M受体拮抗剂
一)茄科生物碱类M受体拮抗剂 阿托品 Atropine 东莨菪碱 Scopolamine 樟柳碱 Anisodine 山莨菪碱 Anisodamine
硫酸阿托品 Atropine Sulphate 结构特点: 1、托品(莨菪醇)和托品酸(莨菪酸)酯化 2、莨菪醇:氢化吡咯+哌啶; 2个C*,内消旋化,无旋光性 3、莨菪酸:1个C*,2个光学异构体
天然:S-(-)-托品酸 • 托品酸在分离提取过程中极易发生消旋化, 故 Atropine为外消旋体。 • 左旋体抗M胆碱作用比消旋体强2倍 • 左旋体的中枢兴奋作用比右旋体强8~50倍,毒性更大 • 所以临床用更安全、也更易制备的外消旋体。
化学性质: 1、叔胺:碱性较强,pKa为9.8,水溶液可使酚酞呈 红色 2、酯键:pH3.5-4.0稳定,碱性下分解成莨菪醇和 消旋莨菪酸 3、Vitali反应
茄科生物碱类中枢作用:氧桥,羟基 东莨菪碱 Scopolamine 阿托品 Atropine 樟柳碱 Anisodine 山莨菪碱 Anisodamine
东莨菪碱的半合成类似物 支气管 胃肠道
二)合成M受体拮抗剂 由阿托品结构改造得到:叔胺类和季胺类 药效基本结构:氨基乙醇酯 酰基上的大基团:阻断M受体功能 合成M受体拮抗剂的结构通式
合成M受体拮抗剂的构效关系 苯环或碳环 叔胺或季胺取代基:甲基乙基丙基异丙基或形成杂环 酯基醚也可去掉 n=2 -OH-H-CH2OH-CONH2
格隆溴铵 奥芬那君 丙环定 托特罗定 托吡卡胺 异丙碘铵
发展方向: M受体亚型选择性拮抗剂 哌仑西平 Pirenzepine 替仑西平 Telenzepine M1,治疗胃及十二指肠溃疡,慢性阻塞性支气管炎
奥腾折帕 Otenzepad 喜巴辛 Himbacine M2 ,窦性心动过缓,心传导阻滞
索非那新 Solifenacin 达非那新 Darifenacin M3 ,尿频、尿失禁
二、 N受体拮抗剂 nicotinic receptor antagonists • N1受体拮抗剂,神经节阻断剂,在交感和副交感神经节选择性拮抗N1受体,阻断神经冲动在神经节中的传递,主要呈现降低血压的作用,现多被其他降压药取代。 • N2受体拮抗剂,神经肌肉阻断剂,与骨骼肌神经肌肉接头处的运动终板膜上的N2受体结合,阻断神经冲动在神经肌肉接头处的传递,导致骨骼肌松弛。临床用作麻醉辅助药。
神经肌肉阻断剂neuromuscular blocking agents • 去极化型肌松药,与N2受体结合并激动受体,使终板膜及邻近肌细胞膜长时间去极化,阻断神经冲动的传递,导致骨骼肌松弛。 • 非去极化型肌松药,和乙酰胆碱竞争,与N2受体结合,因无内在活性,不能激活受体,但是又阻断了乙酰胆碱与N2受体的结合及去极化作用,使骨骼肌松弛,因此又称为竞争性肌松药。可给予抗胆碱酯酶药逆转。 • 双重作用
生物碱类N受体拮抗剂 • 氯筒箭毒碱 • 四氢异喹啉类N受体拮抗剂 • 苯磺阿曲库铵 • 甾类N受体拮抗剂 • 泮库溴铵
一、生物碱类:氯筒箭毒碱 结构特点: 1)双季胺结构 2)季胺N相隔10~12 个原子 3)N原子处在氢化异 喹啉环上并连有苄基 • 临床上第一个非去极化型肌松药,作用较强,但毒副作用大,已少用。
二)季胺类 “软药” 苯磺阿曲库铵 Atracurium Besylate • 避免了对肝、肾代谢的依赖性,解决了其它神经肌肉阻断剂应用中的一大缺陷-蓄积中毒问题。 • 作用强度约为氯筒箭毒碱的1.5倍。 • 起效快(1~2 min),维持时间短(约半小时),不影响心、肝、肾功能,无蓄积性,是比较安全的肌松药。
软药(soft drugs):指本身具有治疗作用的药物,能根据预见的代谢途径和可控制的速度进行代谢分布,在发挥它的治疗作用后即代谢为无毒物质排出体外的药物。与之相对的是硬药。 硬药(Hard drugs):指具有发挥药物作用所 必需的结构特征的化合物,该化合物在生物体内 不发生代谢或转化,可避免产生某些毒性代谢产 物。
Atracurium 的主要代谢方式:a: Hofmann消除反应 b: 酯水解反应
Atracurium的同型药物 Atracurium分子结构中有4个手性中心,以1R-cis,1R -cis的顺苯磺阿曲库铵(Cisatracurium Besilate)活性最强,为Atracurium Besilate的3倍,无引起组胺释放和心血管副作用,已用于临床。










![Nervous System [humans]](https://cdn1.slideserve.com/2761942/nervous-system-humans-dt.jpg)