Download
1 / 52
530 likes | 718 Views
Il file PDB. Il file PDB. http://www.rcsb.org/pdb. Esempio: Deossiemoglobina umana (1a3n).

E N D
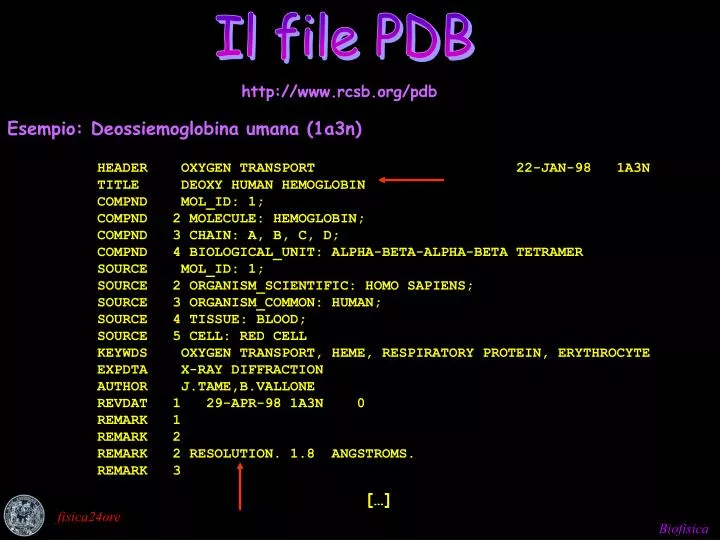
Il file PDB Il file PDB http://www.rcsb.org/pdb Esempio: Deossiemoglobina umana (1a3n) HEADER OXYGEN TRANSPORT 22-JAN-98 1A3N TITLE DEOXY HUMAN HEMOGLOBIN COMPND MOL_ID: 1; COMPND 2 MOLECULE: HEMOGLOBIN; COMPND 3 CHAIN: A, B, C, D; COMPND 4 BIOLOGICAL_UNIT: ALPHA-BETA-ALPHA-BETA TETRAMER SOURCE MOL_ID: 1; SOURCE 2 ORGANISM_SCIENTIFIC: HOMO SAPIENS; SOURCE 3 ORGANISM_COMMON: HUMAN; SOURCE 4 TISSUE: BLOOD; SOURCE 5 CELL: RED CELL KEYWDS OXYGEN TRANSPORT, HEME, RESPIRATORY PROTEIN, ERYTHROCYTE EXPDTA X-RAY DIFFRACTION AUTHOR J.TAME,B.VALLONE REVDAT 1 29-APR-98 1A3N 0 REMARK 1 REMARK 2 REMARK 2 RESOLUTION. 1.8 ANGSTROMS. REMARK 3 […] fisica24ore
tipo di atomo tipo di amminoacido coordinate X Y Z ATOM 1 N VAL A 1 10.720 19.523 6.163 1.00 21.36 N ATOM 2 CA VAL A 1 10.228 20.761 6.807 1.00 24.26 C ATOM 3 C VAL A 1 8.705 20.714 6.878 1.00 18.62 C ATOM 4 O VAL A 1 8.164 20.005 6.015 1.00 19.87 O ATOM 5 CB VAL A 1 10.602 22.000 5.966 1.00 27.19 C ATOM 6 CG1 VAL A 1 10.307 23.296 6.700 1.00 31.86 C ATOM 7 CG2 VAL A 1 12.065 21.951 5.544 1.00 31.74 C ATOM 8 N LEU A 2 8.091 21.453 7.775 1.00 16.19 N ATOM 9 CA LEU A 2 6.624 21.451 7.763 1.00 17.31 C ATOM 10 C LEU A 2 6.176 22.578 6.821 1.00 18.55 C ATOM 11 O LEU A 2 6.567 23.730 7.022 1.00 18.72 O ATOM 12 CB LEU A 2 6.020 21.707 9.129 1.00 18.34 C ATOM 13 CG LEU A 2 6.386 20.649 10.198 1.00 17.39 C ATOM 14 CD1 LEU A 2 5.998 21.119 11.577 1.00 17.99 C ATOM 15 CD2 LEU A 2 5.730 19.337 9.795 1.00 16.96 C ATOM 16 N SER A 3 5.380 22.237 5.852 1.00 15.02 N ATOM 17 CA SER A 3 4.831 23.237 4.928 1.00 16.59 C ATOM 18 C SER A 3 3.725 24.027 5.568 1.00 14.84 C ATOM 19 O SER A 3 3.095 23.717 6.591 1.00 14.40 O ATOM 20 CB SER A 3 4.308 22.429 3.727 1.00 16.47 C ATOM 21 OG SER A 3 3.076 21.786 3.991 1.00 14.91 O … fisica24ore
1a3n catena A C O N S EME Fe fisica24ore
RASMOL v 2.7 http://www.umass.edu/microbio/rasmol/index2.htm fisica24ore
Tecniche computazionali TECNICHE COMPUTAZIONALI L’utilizzo complementare di tecniche di tipo sperimentale e di tipo computazionale è l’approccio ottimale per lo studio dei sistemi e dei processi biologici. Questa considerazione riguarda in particolare gli aspetti strutturali del problema, ovvero la conoscenza della conformazione, o variazione di conformazione, di una molecola biologica in relazione alla sua attività. fisica24ore
risoluzione spaziale misure ad alta risoluzione di strutture molecolari sono possibili solo per sistemi relativamente rigidi risoluzione energetica analisi delle energie di interazione atomica difficoltosa risoluzione temporale i primissimi eventi dei processi biologici sono di difficile misurazione Limiti delle tecniche sperimentali fisica24ore
Limiti delle tecniche computazionali Sistemi biomolecolari troppo complessi meccanica classica con funzioni di interazione semi-empiriche per descrivere le interazioni tra gli atomi di un sistema molecolare Simulazione del comportamento di un sistema molecolare su un computer solo un numero limitato (<NA) atomi o di gradi di libertà (di solito 102-105 atomi), per un limitato periodo di tempo (102-104 picosecondi) può essere simulato piccoli sistemi, con tempi di rilassamento brevi Campionatura limitata dello spazio delle conformazioni di una macromolecola utilizzo dei dati sperimentali per restringerlo fisica24ore
importanza della COMPLEMENTARITA’ DELL’APPROCCIO TEORICO-SPERIMENTALE fisica24ore
Alcune Applicazioni • In primis: conoscere la struttura tridimensionale a risoluzione atomica della molecola per comprendere, spiegare, e a volte anche modificare ed utilizzare, la sua attività biologica. fisica24ore
Monitorare i cambiamenti strutturali indotti su peptidi o proteine da parte di MEMBRANE BIOLOGICHE, i quali sembrano essere fondamentali per il riconoscimento con il recettore o per oltrepassare la fase lipidica e raggiungere zone altrimenti inaccessibili. fisica24ore
Effettuare MUTAZIONI puntiformi, che possono fornire indicazioni utili per il riconoscimento del sito attivo o di strutture indispensabili all'attività della molecola o dirette ad una certa funzione. fisica24ore
Studiare le variazioni conformazionali provocate dall’interazione della proteina con uno o più LIGANDI, la quale fornisce l’attivazione (o inattivazione) necessaria per compiere la propria funzione biologica (o per impedirla). fisica24ore
Comprendere il processo di FOLDING delle proteine, ovvero il meccanismo di ripiegamento con cui raggiungono la confomazione biologicamente attiva. fisica24ore
Applicazioni FARMACOLOGICHE: viene fornita un’indicazione specifica, o quanto meno restrittiva, della struttura opportuna in funzione del bersaglio del farmaco. In questo campo, la costruzione di strutture calibrate permette di ridurre la ricerca ad un ristretto raggio d’azione. fisica24ore
Modellizzazione molecolare Modellizzazione Molecolare fisica24ore
Ipotesi termodinamica di Anfinsen (per proteine a singolo dominio) • L’informazione codificata nella sequenza amminoacidica di una proteina determina completamente la sua struttura nativa • Lo stato nativo è il minimo assoluto dell’energia libera della proteina fisica24ore
Diagramma di flussodella modellizzazioneproteica Sequenza proteica Dati sperimentali Allineamento multiplo di sequenza Ricerca nelle banchedati Assegnazione dei domini Proteina omologa nella banca dati PDB? Predizione della struttura secondaria Predizione del fold No Analisi della famiglia del fold E’ stato predetto un fold? Sì Sì Allineamento delle strutture secondarie Modellizzazione comparativa Allineamento della sequenza alla struttura No Predizione della struttura terziaria Modello tridimensionale della proteina fisica24ore
Modellizzazione comparativa (o per similarità di sequenza) Permette di costruire la struttura tridimensionale di una proteina sulla base della SIMILARITÀ DI SEQUENZA con un’altra proteina di struttura NOTA che viene usata come STAMPO. fisica24ore
* aa identici . aa simili Passi fondamentali 1. Allineamento di sequenza con la/le proteina/e “stampo” fisica24ore
2. Costruzione dello scheletro fisica24ore
3. Inserimento delle catene laterali fisica24ore
4. Inserimento dei loop corrispondenti a “buchi” nell’allineamento fisica24ore
5. Ottimizzazione del modello Regolarizzazione di legami, angoli e torsioni Eliminazioni di clash strutturali Minimizzazione energetica fisica24ore
6. Controllo della qualità del modello fisica24ore
Meccanica molecolare Meccanica molecolare Comprende la DINAMICA MOLECOLARE, la RICERCA DELL’ENERGIA CONFORMAZIONALE, e il RICONOSCIMENTO MOLECOLARE (DOCKING). fisica24ore
Ad ogni conformazione molecolare è associata un’ENERGIA fisica24ore
La forma più semplice dell’energia potenziale di una molecola è : ENERGIA pot = Energia di ALLUNGAMENTO dei legami + Energia di PIEGAMENTO degli angoli di legame + Energia di TORSIONE degli angoli diedri + Energia delle interazioni di NON-LEGAME: repulsioni steriche, interazioni di Van der Waals, interazioni elettrostatiche La meccanica molecolare considera gli atomi come sfere e i legami come molle. Forma equazione + parametri = force field fisica24ore
Ricerca dell’energia conformazionale Ricerca dell'energia conformazionale di una molecola Ipotesi termodinamica di Anfinsen (per proteine a singolo dominio) • L’informazione codificata nella sequenza amminoacidica di una proteina determina completamente la sua struttura nativa • Lo stato nativo è il minimo assoluto dell’energia libera della proteina fisica24ore
La superficie dell'energia libera configurazionale di una proteina è tipicamente "rugosa", poichè esistono molti stati metastabili, alcuni dei quali hanno un'energia molto vicina al minimo globale (problema della ricerca del minimo assoluto) fisica24ore
minimo globale dell'energia potenziale risultante delle forze nulla equilibrio stato più popolato Conformazione energeticamente preferita fisica24ore
Minimizzazione dell’energia Minimizzazione dell'energia Minimizzare l'energia potenziale di una molecola significa trovare un percorso (costituito dalle variazioni dei gradi di libertà intramolecolari) che conduca da una conformazione iniziale alla conformazione a minima energia più vicina (MINIMO LOCALE), usando il minor numero di calcoli possibile. fisica24ore
Strategie di ricerca del minimo globale • Campionamento energetico sistematico: L’energia viene campionata ad intervalli regolari sull’intera estensione di ciascun grado di libertà (tipicamente rotazioni dei legami). fisica24ore
Annealing simulato: riscaldamento ad alta temperatura che modifica la struttura superando le barriere energetiche, seguito da un raffreddamento lento. fisica24ore
Ricerca casuale: campionamento che genera strutture in maniera casuale, le quali vengono poi minimizzate • Dinamica molecolare: superamento delle barriere energetiche conformazionali (più basse dell'annealing) e analisi della stabilità strutturale. fisica24ore
Dinamica molecolare Dinamica molecolare Permette lo studio di processi dinamici complessi che avvengono nei sistemi biologici. Studia sia transizioni conformazionali che vibrazioni locali, ad esempio: stabilità delle proteine variazioni conformazionali folding proteico trasporto ionico fisica24ore
Calcola la TRAIETTORIA di un sistema molecolare = la configurazione molecolare in funzione del tempo, ovvero come variano nel tempo le posizioni, le velocità e le accelerazioni degli atomi della molecola. fisica24ore
La traiettoria è generata da integrazioni simultanee dell’ equazione del moto di Newton Fi = mi ai per tutti gli atomi del sistema molecolare Tenendo presente che la forza si può esprimere come gradiente dell'energia potenziale: Fi = - dV/dri si combinano le due equazioni e si ottiene: - dV/dri = mi d2ri/dt2 che collega la derivata dell'energia potenziale alle variazioni di posizione in funzione del tempo ed è quella che viene integrata. fisica24ore
Perciò per calcolare una traiettoria c'è bisogno: • delle posizioni iniziali degli atomi (coordinate atomiche) • delle velocità iniziali • delle accelerazioni 1. le posizioni inziali ri si ricavano da strutture sperimentali (cristallografia raggi X, NMR ecc.) o ottenute con modeling; 2. le velocità iniziali vi si ottengono dalla distribuzione delle velocità ad una data temperatura; 3. le accelerazioni sono determinate dal gradiente dell'energia potenziale. fisica24ore
Le posizioni e le velocità iniziali (t = 0) determinano le posizioni e le velocità a tutti gli altri tempi t. In pratica si considerano intervalli di integrazione finiti Dt.Dt tipicamente va da 0.1 a 10 fs per i sistemi molecolari una simulazione di 100 ps coinvolge 105-106 intervalli di integrazione. fisica24ore
Riscaldamento lento alla temperatura di simulazione Riequilibrazione dell’energia tra gli atomi Energia cinetica totale del sistema Distribuzione iniziale di velocità vi Sistema allo zero assoluto Temperatura di simulazione desiderata fisica24ore
Protocollo di simulazione Struttura iniziale rimuove interazioni di Van der Waals forti che porterebbero a distorsioni locali Minimizzazione dell’ energia nel caso si usi un solvente esplicito, aggiungere le molecole d’acqua Solvatazione della proteina Minimizzazione dell’ energia in presenza del solvente per equilibrarlo con la struttura si lancia la MD con velocità iniziali a bassa temperatura nuove velocità riassegnate periodicamente a T leggermente più alta e così via fino al raggiungimento della T di simulazione desiderata. Fase di riscaldamento la simulazione prosegue finchè sono stabili nel tempo la struttura, la pressione, la temperatura (si riscalano le velocità), l'energia Fase di equilibrazione Fase di simulazione vera e propria fisica24ore
Analisi dei risultati Analisi dei risultati Campionamento periodico di coordinate (e velocità) Calcolo dell’energia potenziale media in funzione del tempo Calcolo della differenza con la struttura di partenza in funzione del tempo Calcolo della superficie accessibile al solvente e del raggio di girazione, in funzione del tempo Calcolo della struttura media fisica24ore
Riconoscimento molecolare Riconoscimento molecolare E’ il punto di partenza per quasi tutti i processi biologici. Le molecole interagiscono in una maniera altamente specifica: modello CHIAVE-SERRATURA(Fisher e Ehrilch) fisica24ore
La complementarità geometrica e chimica fra piccole molecole biologiche (LIGANDI) e le strutture dei loro bersagli macromolecolari (RECETTORI) gioca un ruolo molto importante all’interno dei processi biologici. fisica24ore
Sviluppo di farmaci Elemento chiave: scoperta di composti guida nuovi e innovativi Composto guida = composto che mostra affinità per un dato recettore, che ha attività biologica e che può essere strutturalmente modificato per migliorare la bioattività fisica24ore
tempi per lo sviluppo di un nuovo farmaco è di grande importanza l’identificazione RAPIDA E AFFIDABILE di ligandi ad alta affinità Tempi per lo sviluppo di un nuovo farmaco • Ricerca del composto guida (1-2 anni) • Ottimizzazione del composto guida (1-2 anni) • Saggi di attività in vitro e in vivo (1-2 anni) • Test tossicologici (1-3 anni) • Test per la sicurezza sull’uomo (1 anno) • Test per l’efficacia sull’uomo (1-2 anni) Tempo totale per lo sviluppo di un nuovo farmaco: 6-12 anni Costo totale: circa $ 500 000 000 fisica24ore
Screening sperimentale: test in vitro di grandi librerie di composti. Ignora, in genere, le proprietà strutturali del recettore Metodi computazionali detti Rational Design: si basano su informazioni strutturali del recettore e/o del ligando Ricerca di nuovi composti guida fisica24ore
De Novo Design I nuovi compostivengono generati nel sito di legame a partire da atomi o frammenti preposizionati nel sito e che successivamente vengono trasformati in molecole intere da softwares specifici. Screening Virtuale Librerie di molecole (esistenti o ipotetiche) vengono analizzate cercando ligandi con caratteristiche in accordo con i requisiti del sito di legame Docking Metodi computazionali Struttura 3D del recettore NON nota Struttura 3D del recettore nota Structure Based Drug Design QSAR (Quantitaive Structure-Activity Relationship) Stabilisce una relazione tra la struttura molecolare e l’attività biologica di una serie di composti attivi. Predice la attività e la affinità di composti non noti dall’analisi delle loro similitudini e differenze strutturali, fornendo informazioni sui requisiti strutturali del recettore. fisica24ore
Docking Predice la struttura 3D di complessi proteina-ligando. Trova il corretto modo di legame di un composto, tramite il campionamento dello spazio conformazionale nel sito di legame, attraverso la valutazione di funzioni che stimano l’energia di ogni combinazione confomazionale ligando-recettore. • Tali funzioni valutano: • Complementarità fra superficie • Energia libera di solvatazione • Interazioni elettrostatiche e idrofobiche fisica24ore