Download

1 / 19
190 likes | 320 Views
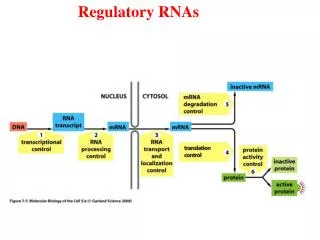
Identifying and classifying functional small RNAs from pine. Ryan Morin BC Genome Sciences Centre (presenting research conducted in the lab of Dr. Peter Unrau). Small RNAs in the land plants. MicroRNA Biogenesis. MicroRNA (miRNA) transcripts can be mono- or polycistronic

E N D
Identifying and classifying functional small RNAs from pine Ryan Morin BC Genome Sciences Centre (presenting research conducted in the lab of Dr. Peter Unrau)
MicroRNA Biogenesis • MicroRNA (miRNA) transcripts can be mono- or polycistronic • Each miRNA derives from its own hairpin foldback region of the primary miRNA transcript • A number of processing steps are required to form the mature miRNA sequence • The final mature miRNA is bound to a complex of proteins known as RISC
MicroRNAs are post-transcriptional gene regulators • MiRNAs are short (18-24nt) RNAs that are derived from larger precursor transcripts • Mature miRNAs hybridize to near-complementary sites within their target messenger RNAs • Messenger RNAs hybridized to microRNAs are enzymatically degraded, preventing translation
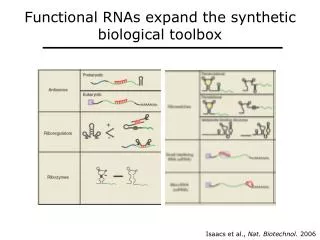
siRNAs can be post- or pre-transcriptional gene regulators • siRNA precursors are also processed by dicer (plants have multiple Dicer paralogs) • Many active mature siRNAs are produced from the primary duplex • Mature siRNAs can elicit post-transcriptional regulation by a microRNA-like mechanism • SiRNAs can also direct methylation of histones or DNA, altering chromatin state (so-called heterochromatin siRNAs)
Purification, adaptor ligation and amplification of small RNAs Extract sequences in desired size range Extract Total RNA Pine Rice smRNA ligate 3’ DNA adaptor ligate 5’ RNA adaptor biotin Gel Purify (remove un-ligated sequences) RT-PCR & PCR amplification Library Identification Tag (8mer) Random 10mer biotin 454 sequencing and amplification primer sequences Forward PCR Primer sequence Reverse PCR Primer complementary sequence 454 sequencing and amplification primer sequences
Rice and Pine sequences produced by 454 SBS technology • 142,493 reads from pine, 11,436 from rice • Small RNA sequence was extracted by identifying flanking adaptor sequences • Random tag sequence was used to separate PCR-amplified constructs from multiple occurrences of the same small RNA species • There were 58,466 unique sequences from pine and 8,615 from rice • Most common sequence was observed 1168 times in pine and 89 times in rice
Positional annotation of small RNAs and identification of conserved pine small RNAs • Alignment of both of pine small RNA sequences to the rice genome reveals perfectly conserved pine small RNAs • Pine sequences aligning to rRNA or tRNA genes in the rice genome are likely degradation products of pine rRNA/tRNA • Sequences aligning within annotated regions were annotated as rRNA, tRNA, miRNA, repeat-derived siRNA etc.
Positionally-annotated small RNAs show distinct trends in sequence length
Pre-miRNA structure: the ‘gold standard’ for miRNA annotation 1)No more than 4 unpaired nt total (or >2 consecutive unpaired nt) within miRNA/miRNA* region 2) The length of the hairpin must be at least 60nt 3) No more than one asymmetrically unpaired nucleotide 4) The pairing must extend 4nt beyond the mature miRNA • To classify a sequence as a miRNA, one usually has to be convinced it forms a suitable pre-miRNA structure • Knowing the sequence of the mature miRNA helps to separate random hairpins from real pre-miRNAs Rules enforced by the Bartel lab (MIRcheck)
Finding clusters of related sequences • Align each pair of sequences in the database including all known miRNAs (mature miRNA sequences from every plant species in miRBase) • Link sequences in the database if they align over >18 nt with < 4 mismatches • Treating these relationships as a graph, extract all the connected components 1 1 1 1 2 2 1 1 2 3 1 1 2 1 1 1
Putting clusters to use Part 1: Guilt by association • Known miRNA families generally partition into the same cluster (along with un-annotated sequences) • If they are low-quality (i.e. large/diverse) clusters are ignored • sequences can be readily annotated as conserved miRNAs if they share a cluster with a known miRNA
Leveraging EST/cDNA sequences(miRNA annotation Method 1) • Pine small RNAs were also aligned to the publicly available pine EST sequences • miRNAs align to their ‘parent’ transcripts in a distinctive way • We fished for un-annotated small RNA sequences with miRNA-like alignments to pine ESTs • Folding and validation of these ESTs revealed 19 putative novel pine miRNA families miRNA* sequences miRNA sequences
Putting clusters to use part 2: miRNA-like Clusters (method 2) • Clusters of known miRNA families had many similarities which might allow their separation from non-miRNA clusters • High information content • Mean sequence length • Low variance in sequence length • Few indels, relatively few genomic loci • A support vector machine (SVM) classifier was trained on all known microRNA clusters • This classifier demonstrated high specificity (0.96) and modest sensitivity (0.65) • 54 clusters were predicted to be miRNA clusters (all pine-specific) and may represent novel pine microRNA families • 5 of these clusters are confirmed miRNAs based on EST folding
Putting clusters to use, part 3: miRNA/miRNA* pairs (method 3) • Maturation of a miRNA often leads to two small RNA molecules: the miRNA and its semi-complementary partner (miRNA*) • Many of the clusters with small RNAs that align in forward/reverse pairs contained known microRNAs • Some of these clusters contained sequences that aligned to ESTs, which could be folded and checked for good pre-miRNA structures • This method revealed an additional 24 clusters of novel miRNA sequences
Mean information content for clusters annotated as microRNAs
Summary • Some of the 24nt heterachromatin siRNAs (including repeat-derived siRNAs) are conserved between gymnosperms and angiosperms • Pine small RNA sequences are rich in conserved microRNAs • Various methods, including sequence clustering, RNA folding and machine learning, reveal many putative novel pine miRNA families
Acknowledgements Thanks: Peter Unrau (SFU, MBB) Cenk Sahinalp & his lab (SFU, CS) Gozde Cozen (SFU, CS) Alex Ebhardt (SFU, MBB) Elena Dolgosheina (SFU, MBB) Matt Hickenbotham (WashU) Vincent Magrini (WashU) The VanBUG Dev team Other VanBUG sponsors: IBM, Genome BC