Download

1 / 35
350 likes | 540 Views
Global Development Meeting the Needs in Japan. Dr David Jefferys Senior Vice President Global Regulatory, Healthcare Policy Department Eisai Europe Ltd Thursday, 9 th June 2011. The Pharmaceuticals and Medical Devices Agency (PMDA) was established in April 2004

E N D
Global DevelopmentMeeting the Needs in Japan Dr David Jefferys Senior Vice President Global Regulatory, Healthcare Policy Department Eisai Europe Ltd Thursday, 9th June 2011
The Pharmaceuticals and Medical Devices Agency (PMDA) was established in April 2004 • Incorporated Administrative Agency of the MHLW
Functions • Integrated Regulatory Agency • Pharmaceutical Affairs Law • GMP inspections collaboration with the prefecture administrations
Previous Controls PMDEC Pharmaceuticals and Medical Devices Evaluation Centre OPSR/Kiko Organisation for Pharmaceutical Safety and Research (RD promotion. Adverse event relief funds, clinical trial appraisals added in 1997 and conformity audit) JAAME Japan Association for the Advancement of Medical Equipment (equivalency review for medical devices 1995)
Unique Functions • Advance health effect relief services • Provides medical expenses, disability pension and bereavement pensions for sufferers of adverse health effects, including adverse reactions to drugs and biological product-derived infections • Provides health allowances to SMON (subacute myelo optico-neuropathy) patients and HIV positive and AIDS patient • Research and Development Promotion Services (April 2005) - Innovative pharmaceuticals and medical devices - Orphan drugs
Japanese Regulatory AuthoritiesRoles and Responsibilities • Ministry of Health, Labour & Welfare (MHLW) - makes all final decisions based on recommendations of PMDA
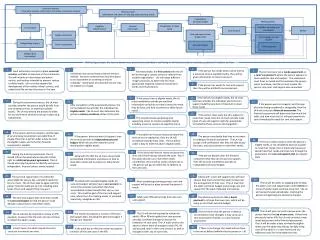
Office of General Affairs Office of Planning & Co-ordination Senior Councillor Office of Relief Funds PMDA Structure Office of Review Administration Office of New Drug 1 Office of New Drug II Chief Executive Office of New Drug III Director, Center for Product Evaluation Priority Review Director Office of Biologics Executive Director Office of OTCs / Generic Evaluation Associate Centre Directors Office of Medical Devices Office of Conformity Auditor Office of Safety Division Chief Safety Officer Office of Compliance Standards Office of R&D Promotion
When are CTNs required? 1. Drug with new active ingredient 2. Drug with new route of administration ( excluding bioequivalence studies) 3. New combination drugs, drugs with new indications or dosage & administration ( excluding bioequivalence studies) 4. Drugs with same active ingredient as drugs with new ingredient for which the have re-examination period has not been completed ( excluding bioequivalence studies) 5. Biological products 6. Drugs manufactured by recombinant DNA technology
Regulatory requirements for a Clinical Trial Notification (CTN) • CTN’s are required for all stages of clinical development • No formal approval is given • package is reviewed • issued raised by authority must be addressed • Study can begin 31 days after submission • contract can be made with institute/hospital • For second CTN and thereafter, time reduced to 14 days • Trial drug can be imported when CTN is submitted. Drug can be supplied to investigator on day 31
Japanese Regulatory AuthoritiesRoles and Responsibilities • Committee on Drugs - First Committee responsible for approval/non-approval of drugs not listed in JP and designation of re-examination term for these drugs namely “drugs which are not handled by the Second Committee on Drugs” - Second Committee responsible for antibacterials, chemotherapeutics, anti-cancer drugs, blood preparations and biological products not listed in JP and designation of re-examination term for these drugs • Executive Committee
Formal Consultations (2-3 hours plus) • Pre-Phase I Consultation : rationale for initiation of Phase I • Pre-early phase II • Post -Phase II : rationale for progression to Phase III / rationale for bridging • Pre-JNDA : guidance on the suitability, inclusion and presentation of clinical data for the J-NDA • Additional consultation : opportunity for continuing discussions from another consultation category
New Agency - User Fee for Review • New drug submission Current ~ £140k • Abridged ~ £70k applications • Target Review times (see website)
Issues to consider for the Japanese population • Pharmacogenetic differences (polymorphisms) • Pharmacodynamic differences • Receptor sensitivity • Diet/effect of food • Dose tolerance • Tolerance/updating of adverse events/reactions • Comparator therapy/interventions
Issues to consider for the Japanese population, contd…. • Prevalence and nature of the disease • Relevance of historical comparisons • Differences in comparator therapy • Different availability of medicines • Differences in healthcare delivery • Cultural differences – social support • GCP – Informed consent
Global Drug Development • Do you need data in Japanese patients - (for a successful file) YES • How do you achieve this: • global protocol with Japanese centres • Japanese study and bridge to US/EU data • ICH E5 (Ethnic Factors in the Acceptability of Foreign Clinical data), Iyakushin Notification 672, 11 August 1998 • Consider: • gold standard in Japan • end points, surrogate markers • disease definition, local guidelines
Phase I • Japan requires a full/appropriate phase I programme • Default position • Agree with PMDA what could be covered by EU/US data • For potential global development projects conduct early Japanese phase I studies
Pre-Phase 1 Consultation • Recommended (industry’s perspective) • Rationale for initiation of Phase I • Example topics for discussion may include - pre-clinical studies required to progress to Phase I - subjects in Phase I - volunteers or patients - dosing regime and dose escalation - utilisation of overseas Phase I data - appropriateness of explanatory documents for informed consent
Phase II Development • Japanese phase II B study, then can consider bridging study for the phase III programme to EU/US data • How to handle adaptive (seamless) designs? • If phase I and II A in Japanese subjects then could go directly to a Japanese phase III study (influenced by stage and extent of development outside Japan)
Post- Phase II Consultation • Essential (industry’s perspective) • Rationale for progression to Phase III/Rationale for bridging • Example topics for discussion may include • evaluation of dose-ranging data • rationale for dose selection • selection of comparator products • clinical endpoints and evaluation criteria • necessity of investigating drug interactions • use of foreign clinical data
Phase III Development Assume a Japanese phase III study will be required Exceptions: • bridging from phase II B data • oncology products (conditional approvals) • combined study (multi-national) adequately powered (destructive analysis) • use of overseas Japanese patients (criteria < 4 years outside Japan/1st generation
Key Issues • Comparator therapies • Different availability of medicines (different Gold standards) • Local guidelines • Dosage issues
Pre - JNDA Consultation • Essential (industry’s perspective) • Guidance on the suitability, inclusion and presentation of clinical data for the JNDA • Example topics for discussion may include • assessment of clinical data versus the proposed label • interpretation/ judgement on the success/failure of bridging
Nature and Extent of the Bridging Studies • E5: sensitivity (relevant ethnic sensitivity) • Experience with similar drugs • Extrinsic factors (medical practice etc) • Safety extrapolation (power of the studies)
Legal background of post-approval commitments and conditional authorisations Early Post-marketing Phase Vigilance (EPPV) • Effective since October 1, 2001 • Ensure that the necessary information on proper use of new drug products is provided to medical institutions 2 weeks prior to the delivery of the products to the institutions • Request that medical institutions expeditiously report on the occurrence of serious ADRs • Repeatedly request that medical institutions use the new drug products properly and report on the occurrence of serious ADRs, during the 6 months after delivery of the products • It is fundamentally the duty of medical institutions to disseminate information on proper use within the institutions, and to co-operate with pharmaceutical companies to collect information on serious ADRs (PAL)
Website for PMDA: http://pmda.go.jp
The Innovate 25 Agenda and New Democratic Party Agenda • Importance of pharmaceuticals • New targets, new funding, expansion of PMDA • Encouragement of innovation • Work sharing • Possible mutual recognition systems (see later slide)
The Innovate 25 Agenda and New Democratic Party Agenda • Medical devices (acceptance of international third party evaluation) • Confidentiality agreements with EMEA February 2007 • Exchange of evaluation reports • Paediatrics trilateral exchange with EMA (PDCO) and FDA initiated November 2009
Addressing the Drug Lag • Assessment of the pharmacogenetic differences with the Asian population Japan China Korea (Taiwan) • Working party has been established • Report awaited
Addressing the Drug Lag, contd Potential outcome: • Use of pooled/common data • Mutual exchange/recognition of data • ASEAN initiatives