Download
1 / 1
10 likes | 201 Views
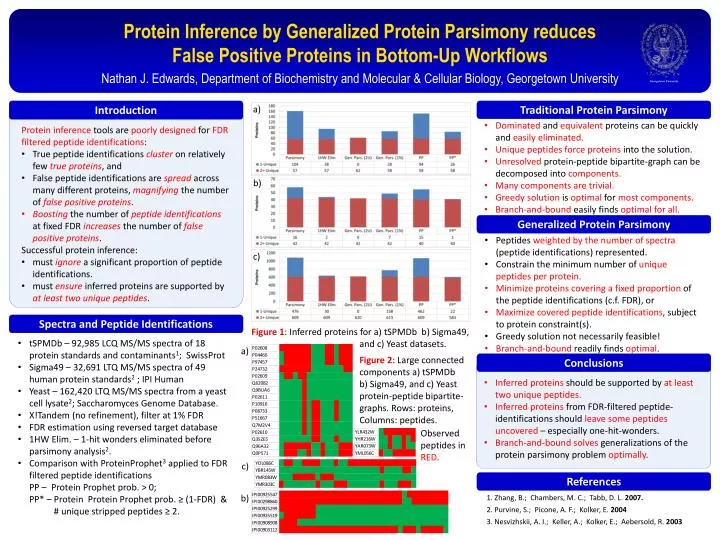
Georgetown University. Protein Inference by Generalized Protein Parsimony reduces False Positive Proteins in Bottom-Up Workflows. Nathan J. Edwards, Department of Biochemistry and Molecular & Cellular Biology, Georgetown University. a). b ). Introduction.

E N D
Georgetown University Protein Inference by Generalized Protein Parsimony reduces False Positive Proteins in Bottom-Up Workflows Nathan J. Edwards, Department of Biochemistry and Molecular & Cellular Biology, Georgetown University a) b) Introduction Protein inference tools are poorly designed for FDR filtered peptide identifications: • True peptide identifications clusteron relatively few true proteins, and • False peptide identifications are spreadacross many different proteins, magnifyingthe number of false positive proteins. • Boostingthe number of peptide identificationsat fixed FDR increasesthe number of false positive proteins. • Successful protein inference: • must ignorea significant proportion of peptide identifications. • must ensureinferred proteins are supported by atleast two unique peptides. c) Figure 1: Inferred proteins for a) tSPMDb b) Sigma49, and c) Yeast datasets. References Spectra and Peptide Identifications a) Traditional Protein Parsimony Generalized Protein Parsimony • Zhang, B.; Chambers, M. C.; Tabb, D. L. 2007. • Purvine, S.; Picone, A. F.; Kolker, E. 2004 • Nesvizhskii, A. I.; Keller, A.; Kolker, E.; Aebersold, R. 2003 • Dominated and equivalent proteinscan be quickly and easily eliminated. • Unique peptides force proteins into the solution. • Unresolved protein-peptide bipartite-graph can be decomposed into components. • Many components are trivial. • Greedy solution is optimal formost components. • Branch-and-bound easily finds optimal for all. • Peptides weighted by the number of spectra (peptide identifications) represented. • Constrain the minimum number of unique peptides per protein. • Minimize proteins covering a fixed proportion of the peptide identifications (c.f. FDR), or • Maximize covered peptide identifications, subject to protein constraint(s). • Greedy solution not necessarily feasible! • Branch-and-bound readily finds optimal. • tSPMDb– 92,985 LCQ MS/MS spectra of 18 protein standards and contaminants1; SwissProt • Sigma49 – 32,691 LTQ MS/MS spectra of 49 human protein standards2; IPI Human • Yeast – 162,420 LTQ MS/MS spectra from a yeast cell lysate2; Saccharomyces Genome Database. • X!Tandem (no refinement), filter at 1% FDR • FDR estimation using reversed target database • 1HW Elim. – 1-hit wonders eliminated before parsimony analysis2. • Comparison with ProteinProphet3 applied to FDR filtered peptide identifications PP – Protein Prophet prob. > 0; PP* – Protein Protein Prophet prob. ≥ (1-FDR) & # unique stripped peptides ≥ 2. Figure 2: Large connected components a) tSPMDb b) Sigma49, and c) Yeast protein-peptide bipartite-graphs. Rows: proteins, Columns: peptides. Observed peptides in RED. c) b) Conclusions • Inferred proteins should be supported by at least two unique peptides. • Inferred proteins from FDR-filtered peptide-identifications should leave some peptides uncovered – especially one-hit-wonders. • Branch-and-boundsolves generalizations of the protein parsimony problem optimally.