Download

1 / 24
240 likes | 260 Views
Explore reconstructing evolutionary history of human gene clusters using algorithms, data preparation, and evaluation methods presented at RECOMB 2008.

E N D
Reconstructing the Evolutionary History of Complex Human Gene Clusters Y. Zhang, G. Song, T. Vinar, E. D. Green, A. Siepel, W. Miller RECOMB 2008 Speaker: Fabio Vandin
Outline • Motivation • Problem definition • Simple algorithm for simple model • SIS algorithm for complex model • Results on human genome • Evaluation of SIS algorithm
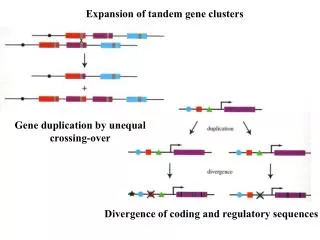
Gene cluster • Group of related genes • Probably formed by duplications: • followed by functional diversification • with deletions, cause human genetic diseases
“History” of a gene cluster? ? 80% 85% 93% 89% 98% Percentage of similarity in self-alignment in the human genome
Dot-plots of self-alignments Human UGT2 cluster
Data Preparation • Atomic segments: • Self-alignment (forward and reverse-complement): pairs A ≈ B • Transitive closure property: A ≈ B, B ≈ C A ≈ C • Maximize each alignment • Collapsible segments:
Computational Problem • Input: sequence of atomic (non-collapsible) segments • Output: (most probable)sequence of events (or the number of events) such that if we unwind these events in the input sequence, we obtain a sequence containing only a single atomic segment
Simple Model • Assumptions: • Only duplications (possibly with reversal and tandem duplications) • No duplication inside the originating region • The most important:
Simple Model (2) • Given assumptions 1 e 2, each event is of “identical” (reversed) regions of consecutive atomic segments ( copied to ) • Problem statement
Simple Model (3) • Candidate definition • Is this definition reasonable?
Simple Model (4) • Algorithm • Analysys
Simple Model: limitations • Assumptions: • “large scale deletions are likely to occur” • “atomic boundary reuses violating assumption are not uncommon” • Same number of events, but multiple way of reconstructing the history • NB: not solved with SIS, but you can assign probability..
Stochastic Model • Event : • Duplication (possibly with reversal) • Deletion (with restrictions) • History: • Target distribution of histories: • is the number of reused atomic boundaries and
Algorithm for Stochastic Model • Sample histories from the target distribution and compute the mean value (of the function we are interested in): • sample from • given , estimate • e.g., gives the number of events • Problem: how to sample from the target distribution?
Sequential Importance Sampling (SIS) • Goal: compute • Target distribution: • Trial distribution: • Sample’s weight: • Output:
SIS for gene cluster history • Duplication
SIS for gene cluster history (2) • Deletion • only without atomic boundary reuse • only if “the atomic segment pair flanking a deletion site appears elsewhere” in the seq.
Application to Human Gene Clusters • human genome assembly hg18 self alignment: 457 duplicated regions • alignments: at least 500 bp, >= 70% identity • segments separated by no more than 500 Kbp • only long and non-trivial regions considered • 165 biomedically interesting clusters (~111 Mbp) • 5 divergence thresholds: GA, OWM, NWM, LG, DOG • 825 combinations of gene cluster and divergence threshold
Application to Human Gene Clusters (4) • “… help prioritize the selection of notably interesting gene clusters for more detailed comparative genomics studies” • “… to compare cluster dynamics in certain lineages to observed phenotypic differences among primates” • “… such sequence data should reveal differences among primate species of possible relevance for selecting species for further biomedical studies”
Evaluation of the SIS Algorithm • Estimate parameters from the human genome for the events (e.g., duplications): • 39% of duplication “reversed”, deletions=2% of duplications • Starting from 500 Kbp sequence, generate 10 genome clusters for N= 10,20,…,100 events
Discussion • Main Limitations: • details of data preparation • choice of the parameters/distributions • evaluation of the SIS algorithm • Future Directions: • include other types of events • understand the stochastic model • how to evaluate a model? • how to evaluate an algorithm?