Download

1 / 42
420 likes | 433 Views
Learn about three types of comparisons - whole genome comparison, gene search, and motif discovery. Understand the importance of alignment in finding shared patterns in genetic data. Recap and prepare for the next quiz.

E N D
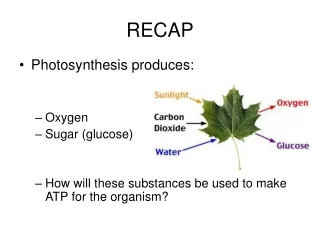
Recap • 3 different types of comparisons • Whole genome comparison • Gene search • Motif discovery (shared pattern discovery)
Agenda • More about Shared Pattern Discovery • Edit Distance • Recap • What you need to know for the next quiz • Alignment • More details • More examples
Shared Pattern Discovery • I have 10 rats that all have green eyes • I have 10 rats that all have blue eyes • What exactly do the 10 rats have in common that give them green eyes?
Shared Pattern Discovery • Multiple Alignment can be used to measure the strength a genomic pattern found in a set of sequences • First, completely align the 10 green-eyed rats • Then, align green-eyed rats with blue-eyed rats • Finally, compare the statistical difference • Initially, this is how genes were pin-pointed
95.2%similar 94.7%similar 99.3%similar 99.2%similar Shared Pattern Discovery 99.4%similar 99.2%similar • Multiple alignment of 10 green-eyed rats 94.5%similar 99.1%similar • Alignment of blue-eyed rat and green-eyed rat
Recap: Exact string matching • Its important to know why exact matching doesn’t work. • Target: CGTACGAC • Pattern: CGTACGTACGTACGTTCA • Problem: Target can NOT be found in the pattern even though there is a near-match • Sequences either match or don’t match • There is no ‘in-between’
Recap: Edit Dist. for Local Search • Question: How many edits are needed to exactly match the target with part of the pattern • Target: CGTACGAC • Pattern: CGTACGTACGTACGTTCA • Answer: 1 deletion • Example of local search • Gene finding
Recap: Edit Dist. for Global Comp. • Question: How many edits are needed to exactly match the ENTIRE target the WHOLE pattern • Target: CGTACGAC • Pattern: CGTACGTACGTACGTTCA • Answer: 10 deletions • Example of global comparison (whole genome comparison)
Quiz coming up! • You need to be able to compute optimal edit distance. • You need to fill-in the table.
T C G A C G T C A 0 1 2 3 4 5 6 7 8 9 T 1 2 1 2 1 2 3 4 3 4 5 5 6 7 6 7 8 G 2 A 3 6 3 4 2 7 5 3 4 6 3 2 5 2 2 3 5 4 3 6 1 5 2 4 3 2 3 2 5 4 1 3 3 2 1 5 2 4 3 3 2 2 1 2 2 3 3 5 4 5 3 4 4 6 3 C 4 G 5 T 6 G 7 C 8 Edit Distance – Dynamic Programming Optimal edit distance forTG and TCG 0 Optimal edit distance for TG and TCGA 1 Optimal edit distance forTGA and TCGA Optimal edit distance forTGA and TCG Final Answer
0 1 2 3 4 5 6 7 8 9 1 0 2 1 1 2 2 3 4 3 5 4 5 6 7 6 8 7 2 1 3 5 6 4 7 2 3 6 3 4 2 5 3 2 3 4 5 2 3 3 5 4 1 2 6 3 2 5 4 1 2 2 3 1 5 3 2 2 2 4 3 3 1 3 2 4 3 2 5 3 6 5 4 3 4 4 5 6 7 8 Edit Distance int matrix[n+1][m+1]; for (x = 0; x <= n; x++) matrix[x][0] = x; for (y = 1; y <= m; y++) matrix [0][y] = y; for (x = 1; x <= n; x++) for (y = 1; y <= m; y++) if (seq1[x] == seq2[y]) matrix[x][y] = matrix[x-1][y-1]; else matrix[x][y] = max(matrix[x][y-1] + 1, matrix[x-1][y] + 1); return matrix[n][m];
This is a gene in the rat genome This is the same gene in the fruit bat This is a totally unrelatedregion of the AIDS virus Why Edit Distances Stinks for Genetic Data? • DNA evolves in strange ways • …TAGATCCCAGATCAGTATTCAAGTTATAC…. • …GATCTCCCAGATAGAAGCAGTATTCAGTCA… • … CCTATCAGCAGGATCAAGTATGTCATACTAC… • The edit distance between rat and virus is smaller thanrat and fruit bat.
Alignment • We need a more robust way to measure similarity • Alignment meets several requirements • It rewards matches • It penalizes mismatches • Different strategies for penalizing gaps • It helps visualize similarity.
Alignment • Two examples • What’s more similar • Seq1 & Seq2, or • Seq3 & Seq4
Alignment • Three steps in the dynamic programming algorithm for alignment • Initialization • Matrix fill (scoring) • Traceback (alignment)
Matrix Fill • For each position, Mi,j is defined to be the maximum score at position i,j • Mi,j = MAX [ Mi-1, j-1 + Si,j (match/mismatch), Mi,j-1 + w (gap in sequence #1), Mi-1,j + w (gap in sequence #2) ]
Matrix Fill • Mi,j = MAX [ Mi-1, j-1 + Si,j (match/mismatch), Mi,j-1 + w (gap in sequence #1), Mi-1,j + w (gap in sequence #2) ] • Si,j = 1 if symbols match, otherwise • Si,j = 0 • w = 0 (no gap penalty)
Matrix Fill • The score at position 1,1 can be calculated. • The first residue in both sequences is a GThus, S1,1 = 1 • Thus, M1,1 = MAX[M0,0 + 1, M1,0 + 0, M0,1 + 0] = MAX[1, 0, 0] = 1.
Tracing Back (Seq #1) A | (Seq #2) A
Tracing Back (Seq #1) A | (Seq #2) A
Tracing back the alignment (Seq #1) TA | (Seq #2) A
Tracing Back (Seq #1) TTA | (Seq #2) A
Tracing Back (Seq #1) GAATTCAGTTA | | || | | (Seq #2) GGA_TC_G__A
Robust Scoring • Mi,j = MAX [ Mi-1, j-1 + Si,j (match/mismatch), Mi,j-1 + w1 (gap in sequence #1), Mi-1,j + w2 (gap in sequence #2) ]
Alignment Scoring Alignment score = 8.4
Alignment Scoring Can you find a better alignment?
Alignment Scoring Alignment score = 7.8
Alignment Scoring • Summary: • We have a way of rewarding different types of matches and mismatches • We have a separate way of penalizing gaps • We could choose not to penalize gaps • if we knew that didn’t affect biological similarity • We could even reward some types of mismatches • if we knew they were still biological similarity
Alignment scoring • Process • Experts (chemists or biologist) look at sequence segments that are known to be biologically similar and compare them to sequence segments that are biologically disimilar. • Use direct observation and statistics to develop a scoring scheme • Given the scoring scheme, develop an algorithm to compute the maximum scoring alignment.
Scoring matrix Gap penalty A C G T A 5 -3 -4 -5 -8 C -3 4 -4 -4 G -4 -4 4 -3 T -5 -4 -3 5 Alignment – Algorithmic Point of View • Align the symbols of two strings. • Maximize the number of symbols that match. • Minimize the number of symbols that do NOT match • Gaps can be inserted to improve alignments. • A scoring system is used to measure the quality of an alignment. • In practice: • Scoring matrices and gap penalties are based on biological knowledge and statistical analysis
Local Alignment and Global Alignment • In Global Alignment the two strings must be entirely aligned (every aligned pair of symbols is scored). • In Local Alignment segments from each string are aligned and the rest of the string can be ignored • Global alignment is used to compare the similarity of entire organisms • Local alignment is used to search for genes
Alignment Scoring Revisited • Given a scoring system, the alignment score is the sum of the scores for each aligned pair of symbols plus the gap penalties Local Alignment Total Score = 15
Alignment - Computer Science Perspective • Given two input strings and a scoring system, find the highest scoring local alignment among all possible alignments. • Fact: The number of possible alignments grows exponentially with the length of the input strings • Solving this problem efficiently was an open problem until Smith and Waterman (1980) designed an efficient dynamic programming algorithm • The algorithm takes O(nm) time where n and m are the lengths of the two input strings
Interesting History • The Smith Waterman algorithm for computing local alignment is considered one of the most important algorithms in computational biology. • However, the algorithm is merely a generalization of the edit distance algorithm, which was already published and well-known in computer science. • Converting the edit distance algorithm to solve the alignment problem is “trivial.” • Smith and Waterman are consider almost legendary for this accomplishment. • It is a perfect example of “being in the right place at the right time.”
D[i][j]=MAX( 0, M[i-1][j-1] + S(i,j), M[i-1][j] + w, M[i][j-1] + w); S(i,j) A C G T A 8 -3 -4 -5 C -3 7 -4 -4 G -4 -4 7 -3 0 A 0 C 0 G 0 C 0 Dynamic programming table T -5 -4 -3 8 w -5 A 0 8 3 0 0 G 0 3 4 C 0 T 0 Smith Waterman Algorithm i -4 -5 j -5