Download

1 / 69
890 likes | 1.6k Views
Homology Modeling. Masa Watanabe University of California - Merced. Part 1 Monday 1:30-3:00pm. Homology Modeling - I (Comparative Modeling). What is it and why do we need it? principles of modeling, applications available Using Swiss-Model preparation, workspace tools, automated-mode

E N D
Homology Modeling Masa Watanabe University of California - Merced
Homology Modeling - I(Comparative Modeling) • What is it and why do we need it? • principles of modeling, applications available • Using Swiss-Model • preparation, workspace tools, automated-mode • refining with manually-adjusted alignments • Model Assessment • using Swiss-Model tools to check model quality • what to look for, what we expect…
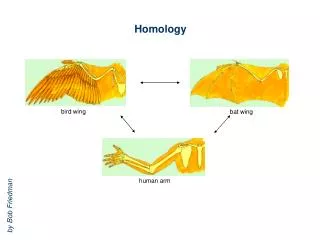
| |..|. ||| ... |..||.|.| | |||..| MHQQVSDYAKVEHQWLYRVAGTIETLDNMSPANHSDAQTQAA | = Identity . | = Homology Why is Homology Modeling? • Given an unknown protein, make an informed guess on its 3D structure based on its sequence: • Search structure databases for homologous sequences • Transfer coordinates of known protein onto unknown MQEQLTDFSKVETNLISW-QGSLETVEQMEPWAGSDANSQTEAY
Why Modeling is Necessary • Current structure determination methods: • NMR – conc. protein solution; limited size + resolution • XRay Crystallography – protein crystals, high resolution • Expensive, slow, difficult (especially membrane proteins!) • Can’t keep up with growth rate of sequence databases: • PDB: 56800 Protein structures (as of 11/10/2009 ) • http://www.pdb.org/ • UniProt Knowledgebase: 512000+ sequences (as of 11/03/2009 ) • http://au.expasy.org/sprot/
High Expectation in Modeling? • At the very best, however, you cannot learn fine details of side-chain conformations, as you can from determining protein structure by X-ray crystallography or NMR. • But homology models can be useful for preliminary exploration, and might also point to useful target residues for chemical analyses of structure, such as site-directed mutagenesis.
Current (Free) Servers & Software • SWISS-MODEL (www) http://swissmodel.expasy.org/ • 3D-Jigsaw (www) http://www.bmm.icnet.uk/servers/3djigsaw/ • ESyPred3D (www) http://www.fundp.ac.be/sciences/biologie/urbm/bioinfo/esypred/ • I-TASSER http://zhang.bioinformatics.ku.edu/I-TASSER/ • WHATIF (www) http://swift.cmbi.ru.nl/servers/html/index.html • CPHmodels 3.0 (www) http://www.cbs.dtu.dk/services/CPHmodels • MODELLER 9v6 (standalone for windows, mac, linux; also web submission) http://www.salilab.org/modeller/ No 1 server in recent CASP7 and CASP8
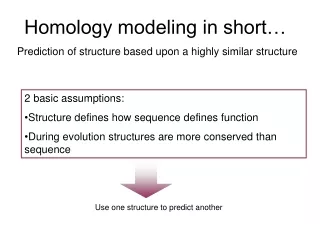
Assumptions & Principles • Increase in sequence identity correlates with increase in structural similarity • RMSD of corea-carboncoordinates for two homologous proteins sharing 50% identity expected at ~1Å (GLY 3.5Å, helix 5Å diameter) • Theoretical models are low resolution, and depend on quality of input alignment!
Sequence alignment is the single most important step in homology modeling.
Important terminology • The protein(s) whose structure(s) is/are already known are referred to as the "reference", "real", or “template” protein(s). • The protein whose structure you are trying to build is called the "model", "unknown", or "target" protein.
Alignment is the limiting step for homology model accuracy No amount of forcefield minimization will put a misaligned residue in the right place ! HOMSTRAD @ CASP4: Williams MG et al. (2001) ProteinsSuppl.5: 92-97
Model Accuracy – Swiss-Model % sequence identity with the submitted sequences (www.expasy.ch/swissmod)
RMSD 1.17Å RMSD 0.41Å RMSD 1.34Å Sidechains Core backbone Loops Sidechains Core backbone Loops Alignment Fold assignment Sidechains Core backbone Loops Alignment Model Accuracy Marti-Renomet al.Annu.Rev.Biophys.Biomol.Struct. 29, 291-325, 2000. HIGH ACCURACY LOW ACCURACY MEDIUM ACCURACY NM23 Seq id 77% CRABP Seq id 41% EDN Seq id 33% X-RAY / MODEL 4/6/03
Swiss-Model steps: Search for sequence similarities BLASTP against EX-NRL 3D Bordoli, etc. (2009) Nature Protocols, 4: 1
Swiss-Model steps: Identity: > 25% Expected model : > 20 resid. Search for sequence similarities Evaluate suitable templates Bordoli, etc. (2009) Nature Protocols, 4: 1
Swiss-Model steps: Search for sequence similarities Evaluate suitable templates Generate structural alignments Select regions of similarity and match in coordinate-space (EXPDB). Bordoli, etc. (2009) Nature Protocols, 4: 1
Swiss-Model steps: Search for sequence similarities Evaluate suitable templates Generate structural alignments Average backbones Compute weighted average coordinates for backbone atoms expected to be in model. Bordoli, etc. (2009) Nature Protocols, 4: 1
Swiss-Model steps: Search for sequence similarities Evaluate suitable templates Generate structural alignments Average backbones Build loops Pick plausible loops from library, ligate to stems; if not possible, try combinatorial search. Bordoli, etc. (2009) Nature Protocols, 4: 1
Swiss-Model steps: Search for sequence similarities Evaluate suitable templates Generate structural alignments Average backbones Build loops Bridge incomplete backbones Bridge with overlapping pieces from pentapeptide fragment library, anchor with the terminal residues and add the three central residues. Bordoli, etc. (2009) Nature Protocols, 4: 1
Swiss-Model steps: Search for sequence similarities Evaluate suitable templates Generate structural alignments Average backbones Build loops Bridge incomplete backbones Rebuild sidechains Rebuild sidechains from rotamer library - complete sidechains first, then regenerate partial sidechains from probabilistic approach. Bordoli, etc. (2009) Nature Protocols, 4: 1
Swiss-Model steps: Search for sequence similarities Evaluate suitable templates Generate structural alignments Average backbones Build loops Bridge incomplete backbones Rebuild sidechains Energy minimize Gromos 96 - Energy minimization Bordoli, etc. (2009) Nature Protocols, 4: 1
Swiss-Model steps: Search for sequence similarities Evaluate suitable templates Generate structural alignments Average backbones Build loops Bridge incomplete backbones Rebuild sidechains Energy minimize Write Alignment and PDB file e-mail results Bordoli, etc. (2009) Nature Protocols, 4: 1
Swiss-Model comparison 3D-Crunch: 211,000 sequences -> 64,000 models Controls: >50 % ID: ~ 1 Å RMSD 40-49% ID: 63% < 3Å 25-29% ID: 49% < 4Å Manual alternatives: Modeller ... Automatic alternatives: SwissModel sdsc1 3djigsaw pcomb_pcons cphmodels easypred # 1 for RMSD and % correct aligned, #2 for coverage Guex et al. (1999) TIBS24:365-367 EVA:Eyrich et al. (2001) Bioinformatics17:1242-1243 (http://cubic.bioc.columbia.edu/eva)
The Swiss-Model Workspace Bordoli, etc. (2009) Nature Protocols, 4: 1 Arnold, etc. (2006), Bioinformatics, 22: 195
Preparation • Automated mode: • Select a template structure with high % identity and average resolution, or let SwissModel choose best one. • Alignment mode: • Obtain multiple alignment between possible templates and your query sequence. • Check alignment – are key motifs correctly aligned? Manually adjust as necessary, using Jalview. • Upload alignment, select template, and submit.
Submit a Model Request Automated Mode
Submit a Model Request Alignment Mode
Submit a Model Request Alignment Mode
Submit a Model Request Alignment Mode
Submit a Model Request Alignment Mode
Hidden Modeling Steps • Automated mode, multiple templates: • Superimposition and optimization of correspondingα-carbonpairs for all template structures • Alignment of all residues of template structures: acceptable alpha-carbon atoms located within3Å radius of mean • All modes: • Framework construction of query protein structure, using coordinates of template structure, or mean coordinate positions if multiple templates. • Building unconserved loop regions / insertions, closing gaps in backbone - uses penta-peptide fragment library derived from all PDB entries of resolution 2Å or better (note: this can fail badly!)
Output • Within a few minutes of submission, your results are returned to the Workspace. • Output Page: • Your model in pdb format (plus simple viewer) • Query to template alignment • Simple assessment graphs • Logging data of the modeling process • Save the model in Swiss-PdbViewer (DeepView) Project format, then open in Swiss-PdbViewer, and color by B-factor. Next Session
Tutorial Exercise 1 • UniProt entry Q11CR8_MESSB (NCBI entry YP_675972) is described as a "Fructose-bisphosphatealdolase class 1, from Mezorhizobium sp. (strain BNC1)” • UniProt Homepage: http://www.uniprot.org/ • NCBI Homepage: http://www.ncbi.nlm.nih.gov/ • It belongs to Pfam (http://pfam.sanger.ac.uk/) PF00274, the Fructose-1,6-Bisphosphate Aldolases (Class 1); this family contains 23 PDB structures. • When we run BlastP on Q11CR8_MESSB against PDB, we get hits to all these structures; %ids range from 48 to 54%, which is reassuringly high. • Let’s try SwissModel in Automated mode: • http://swissmodel.expasy.org/workspace
Tutorial Exercise 2 • Again, let’s try SwissModel in Automated mode for the following: • http://swissmodel.expasy.org/workspace >976347 Human zinc finger homeodomain protein KPFRCEVCNYSTTTKGNLSIHMQSDKH The steps involved in this process may be: First, let’s use the automated mode of Swiss-model to model this protein.
Multiple Sequence Alignment • More information than pairwise alignment • Shows conserved regions in protein family • Critical secondary structure elements • Shows strongly conserved residues and motifs • Structural importance • Functional importance
Alignment Adjustment • Look at alignment used to select template • Check alignment against 3D structure (Jalview) • Secondary structure elements preserved? • Motifs and key residues aligned? • Are gaps in acceptable locations (ie: loops)? • Try to improve alignment manually ... • Or use different alignment application
Template Search Search structure databases for potential homologues, and obtain family information where possible.
Template Search Search structure databases for potential homologues, and obtain family information where possible.