Download

1 / 3
30 likes | 52 Views
Homology ModelingHomology ModelingHomology ModelingHomology Modeling

E N D
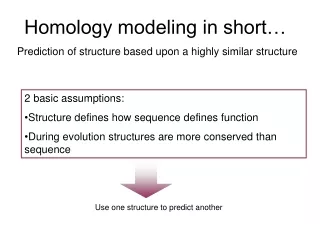
Homology Modeling INQUIRY The three-dimensional structure of a protein is essential to the understanding of its biological function and the design of drug candidates. In recent years, tremendous progress has been achieved in structural biology and a large of number of new macromolecular structures have been determined using experimental methods such as X-ray crystallography, NMR and cryo-EM. Despite the impressive advances, it is still generally difficult and time-consuming to obtain structures for every protein of interest. This is why computational approaches come into play as a powerful alternative and supplement to experimental methods. Homology modeling (also known as comparative modeling) is, to date, the most reliable and well-established computational approach for predicting protein structures. In this method, one or more experimental three-dimensional structures of related homologous proteins are identified and used as templates, based on which an atomic-resolution model of the "target" protein is built from its amino acid sequence. It has been shown that three-dimensional protein structure is evolutionarily more conserved than would be expected on the basis of sequence conservation alone. Thus, even proteins that have diverged appreciably in sequence but still share detectable similarity will also share common structural properties, particularly the overall folding. Protein structure prediction by homology modeling
Profacgen takes advantage of the homology modeling method to help customers predict the three-dimensional structure of proteins of interest. We have extensive experience with the modeling of various monomeric and oligomeric proteins. The resultant structural models are all quality validated and can be used for functional annotation of genes, molecular docking and directing further experimental work such as protein engineering and drug design. Features Multiple-template recognition and selection Accurate sequence alignment with manual corrections Iterative loop modeling and side-chain optimization Prediction of multi-oligomeric proteins Support induced fit effect upon binding of ligand/cofactor Support additional restraints Support incorporation of unnatural amino acids Quality assessment by multiple criteria
Our goal is to predict a structure of protein from its sequence with an accuracy comparable to the best results achieved experimentally. Large-scale automated modeling of protein-coding regions in a genome is also available at Profacgen. We customize the service according to the specific requirements from the customers. Please do not hesitate to contact us for more details about our homology modeling service.