Download

1 / 43
450 likes | 606 Views
Lab 9.3a: Homology Modeling. Boris Steipe boris.steipe@utoronto.ca http://biochemistry.utoronto.ca/steipe Departments of Biochemistry and Molecular and Medical Genetics Program in Proteomics and Bioinformatics University of Toronto. Concepts.

E N D
Lab 9.3a:Homology Modeling Boris Steipe boris.steipe@utoronto.ca http://biochemistry.utoronto.ca/steipe Departments of Biochemistry and Molecular and Medical Genetics Program in Proteomics and Bioinformatics University of Toronto
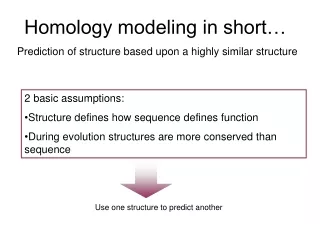
Concepts • Sequence alignment is the single most important step in homology modeling. • Reasons to model need to be defined. • Fully automated homology modeling services perform well. • SwissModel in practice.
Concept 1: Sequence alignment is the single most important step in homology modeling.
What is conserved in structure? E-E.coli [...] IKTRFAPSPTGYLHVGGARTA [...] EQMAKGE----KPRYDGRC [...] AHVSMINGDDGKKLSKRH E-P.putida [...] VRTRIAPSPTGDPHVGTAYIA [...] EQQARGE----TPRYDGRA [...] CYMPLLRNPDKSKLSKRK Q-E.coli [...] VHTRFPPEPNGYLHIGHAKSI [...] TLTQPGKNSPYRDRSVEEN [...] YEFSRL-NLEYTVMSKRK Q-Fly [...] VHTRFPPEPNGILHIGHAKAI [...] FNPKPS---PWRERPIEES [...] WEYGRL-NMNYALVSKRK Q-Human [...] VRTRFPPEPNGILHIGHAKAI [...] HNTLPS---PWRDRPMEES [...] WEYGRL-NLHYAVVSKRK E-Fly [...] VVVRFPPEASGYLHIGHAKAA [...] QRVE----SANRSNSVEKN [...] WSYSRL-NMTNTVLSKRK E-Human [...] VTVRFPPEASGYLHIGHAKAA [...] QRIE----SKHRKNPIEKN [...] WEYSRL-NLNNTVLSKRK E-Yeast [...] VVTRFPPEPSGYLHIGHAKAA [...] DGVA----SARRDRSVEEN [...] WDFARI-NFVRTLLSKRK ATP-Binding | || | || || | QRS E. coli vs. ERS P. putida: ~ 19% ID Many regions are expected to be highly conserved in structure. Some changes should be straightforward to model.
What is conserved in structure? E-E.coli [...] IKTRFAPSPTGYLHVGGARTA [...] EQMAKGE----KPRYDGRC [...] AHVSMINGDDGKKLSKRH E-P.putida [...] VRTRIAPSPTGDPHVGTAYIA [...] EQQARGE----TPRYDGRA [...] CYMPLLRNPDKSKLSKRK Q-E.coli [...] VHTRFPPEPNGYLHIGHAKSI [...] TLTQPGKNSPYRDRSVEEN [...] YEFSRL-NLEYTVMSKRK Q-Fly [...] VHTRFPPEPNGILHIGHAKAI [...] FNPKPS---PWRERPIEES [...] WEYGRL-NMNYALVSKRK Q-Human [...] VRTRFPPEPNGILHIGHAKAI [...] HNTLPS---PWRDRPMEES [...] WEYGRL-NLHYAVVSKRK E-Fly [...] VVVRFPPEASGYLHIGHAKAA [...] QRVE----SANRSNSVEKN [...] WSYSRL-NMTNTVLSKRK E-Human [...] VTVRFPPEASGYLHIGHAKAA [...] QRIE----SKHRKNPIEKN [...] WEYSRL-NLNNTVLSKRK E-Yeast [...] VVTRFPPEPSGYLHIGHAKAA [...] DGVA----SARRDRSVEEN [...] WDFARI-NFVRTLLSKRK ATP-Binding | || | || || | How would sidechain rotamers be modeled? - conserved dihedral angles - preferred rotamers - DEE (Dead End Elimination theorem) for global consistency.
Homology Modeling Issues E-E.coli [...] IKTRFAPSPTGYLHVGGARTA [...] EQMAKGE----KPRYDGRC [...] AHVSMINGDDGKKLSKRH E-P.putida [...] VRTRIAPSPTGDPHVGTAYIA [...] EQQARGE----TPRYDGRA [...] CYMPLLRNPDKSKLSKRK Q-E.coli [...] VHTRFPPEPNGYLHIGHAKSI [...] TLTQPGKNSPYRDRSVEEN [...] YEFSRL-NLEYTVMSKRK Q-Fly [...] VHTRFPPEPNGILHIGHAKAI [...] FNPKPS---PWRERPIEES [...] WEYGRL-NMNYALVSKRK Q-Human [...] VRTRFPPEPNGILHIGHAKAI [...] HNTLPS---PWRDRPMEES [...] WEYGRL-NLHYAVVSKRK E-Fly [...] VVVRFPPEASGYLHIGHAKAA [...] QRVE----SANRSNSVEKN [...] WSYSRL-NMTNTVLSKRK E-Human [...] VTVRFPPEASGYLHIGHAKAA [...] QRIE----SKHRKNPIEKN [...] WEYSRL-NLNNTVLSKRK E-Yeast [...] VVTRFPPEPSGYLHIGHAKAA [...] DGVA----SARRDRSVEEN [...] WDFARI-NFVRTLLSKRK ATP-Binding | || | || || | How would you (or should you even) model indels? - Where should the insertion be placed? - What is the conformation of the new residues? - Which residues should be deleted? - How many additional residues need to change conformation?
Alignment is the limiting step for homology model accuracy No amount of forcefield minimization will put a misaligned residue in the right place ! HOMSTRAD @ CASP4: Williams MG et al. (2001) ProteinsSuppl.5: 92-97
Superposition vs. Alignment The coordinates of two proteins can be superimposed in space. An alignment may be derived from a superposition by correlating residues that are close in space. An optimal sequence alignment may lead to a different alignment ... 1GTR vs 2TS1
TyrRS ERVTLYCGFDPTAdS--LHIGHLATILTMRRFQQAGHRPIALVGGAtgligdpsgkkser | | | ||||| | | | | | 1GTR 26 TTVHTRFPPEPNG-YLHIGHAKSICL--NF---------------GIAqDYKGQCN-- | | ||||| | 2TS1 29 ERVTLYCGFDPTAdSLHIGHLATILT--MR---------------RFQ-QAGHRPI-- TyrRS tlnaketVEAWSARIKEQLgrfldfeadgnpa----------------k--------IKN | | | || | ||| 1GTR 26 ----------------------LRFD-DTnpv----------------keDIEYVESIKN || 2TS1 29 ----------------------ALVG-GAtgligdpsgkksertlnaketVEAWSARIKE TyrRS NYDWIgpldvitflrdvgk----hfsvnymmakesvqsrietgisftefsYMMLQAYDFL | | | | | | | 1GTR 26 DVewl------------gf----hwsgnVRYSSD---------------------YFdql | 2TS1 29 QLgrf------------ldfeadgnpakIKNNYD---------------------WIgpl TyrRS RLYetegCRLQIGGSDQwgnitaGL--------ELIRKTKgearAFGLTIPLV | | | || | || | | | 1GTR 26 hayaie-------------linkglayvdeltpeqireyrgtltqpgknspyrdrsveen 2TS1 29 dvitfl-------------rdvgkhfsvnym----------------------------- TyrRS 1GTR 26 lalfekmraggfeegkaclrakidmaspfivmrdpvlyrikfaehhqtgnkwciypmYDF | 2TS1 29 -------------------------------------makesvqsrietgisftefsYMM TyrRS 1GTR 26 THCISDALEG----ITHSLCTLEFqdnrrlYDWVLDNITipvhPRQYEFSRL 262 2TS1 29 LQAYDFLRLYetegCRLQIGGSDQwgnitaGLELIRKTKgearAFGLTIPLV 223 Superposition vs. Alignment Example: structural vs. sequence alignment between E. coli GlnRS and G. stearothermophilus TyrRS. Although the optimal sequence alignment is not unreasonable (19% ID = 40/212 residues), comparison with the structure shows it is actually wrong for all but 11 residues ! The structure based alignment is quite dissimilar in sequence ( 4.5%ID = 12/265 residues) but the superposition actually matches 39% of residues ( 104/265 ) over the length of the domain.
Inserts may be accomodated in a distant part of the structure Example - a five residue insert Sequence aligment (shows what happened) gktlit nfsqehip gktlisflyeqnfsqehip Structure alignment (shows how it's accomodated) gktlitnfsq ehip gktlisflyeqnfsqehip a-helix
Off by 1, Off by 4 3.8Å • A shift in alignment of 1 residue corresponds to a skew in the modeled structure of about 4 Å (3.8 Å is the inter-alpha carbon distance) • Nothing you can do AFTER an alignment will fix this error (not even molecular dynamics).
Indels (inserts or deletions) • Observations of known similarities in structures demonstrate that uniform gap penalty assumptions areNOT BIOLOGICAL. • Indels are most often observed in loops, less often in secondary structure elements • When they do not occur in loops, there is usually a maintenance of helical or strand properties.
Can we do better with the gap assumption? • Required: position specific gap penalties • One approach: implemented in Clustal as secondary structure masks • Get secondary structure information, convert it to Clustal mask format. (Easy - read documentation !)
Secondary structure from PDB .... (Algorithm ?)
Secondary structure from RasMol .... (DSSP !)
Concept 2: Reasons to model need to be defined.
Use of homology models Biochemical inference from 3D similarity • Bonds • Angles, plain and dihedral • Surfaces, solvent accessibility • Amino acid functions, presence in structure patterns • Spatial relationship of residues to active site • Spatial relationship to other residues • Participation in function / mechanism • Static and dynamic disorder • Electrostatics • Conservation patterns (structural and functional) • Posttranslational modification sites (but not structural consequences!) • Suitability as drug target Don't !
Abuse of homology models • Modelling properties that cannot / will not be verified • Analysing geometry of model • Interpreting loop structures near indels • Inferring relative domain arrangement • Inferring structures of complexes
Databases of Models Don’t make models unless you check first... • Swiss-Model repository • 64,000 models based on 4000 structures and Swiss-Prot proteins • ModBase • Made with "Modeller" - 15,000 reliable models for substantial segments of approximately 4,000 proteins in the genomes of Saccharomyces cerevisiae, Mycoplasma genitalium, Methanococcus jannaschii, Caenorhabditis elegans, and Escherichia coli.
Concept 3: Fully automated services perform well.
TEM HOM TAR Homology Modeling Process PSI-BLAST Search nr (PDB) These are really two queries rolled into one procedure. TAR: Target sequence T-Coffee Align Search: Sequence database similarity search Cinema nr: non-redundant Genbank subset, (with annotated structures) MSA HOM: Homologous sequences SwissModel Model ExPDB TEM: Sequences of homologues with known structure LIG Align: Careful Multiple Sequence Alignment 3D MSA: Multiple Sequence Alignment Model: Generate 3D Model TextEditor Complete ExPDB: Modeling template structure database 3DC Complete: Add ligands, substrates etc. to model Analyse: Interpret and conclude RasMol Analyse PUB: Publish results Consurf PUB
Homology Modeling Software? • Freely available packages perform as good as commercial ones at CASP (Critical Assessment of Structure Prediction) • Swiss Model (see your Integrated Assignment) • Modeller (http://guitar.rockefeller.edu)
Swiss-Model steps: Search for sequence similarities BLASTP against EX-NRL 3D Peitsch M & Guex N (1997) Electrophoresis 18: 2714
Swiss-Model steps: Identity: > 25% Expected model : > 20 resid. Search for sequence similarities Evaluate suitable templates Peitsch M & Guex N (1997) Electrophoresis 18: 2714
Swiss-Model steps: Search for sequence similarities Evaluate suitable templates Generate structural alignments Select regions of similarity and match in coordinate-space (EXPDB). Peitsch M & Guex N (1997) Electrophoresis 18: 2714
Swiss-Model steps: Search for sequence similarities Evaluate suitable templates Generate structural alignments Average backbones Compute weighted average coordinates for backbone atoms expected to be in model. Peitsch M & Guex N (1997) Electrophoresis 18: 2714
Swiss-Model steps: Search for sequence similarities Evaluate suitable templates Generate structural alignments Average backbones Build loops Pick plausible loops from library, ligate to stems; if not possible, try combinatorial search. Peitsch M & Guex N (1997) Electrophoresis 18: 2714
Swiss-Model steps: Search for sequence similarities Evaluate suitable templates Generate structural alignments Average backbones Build loops Bridge incomplete backbones Bridge with overlapping pieces from pentapeptide fragment library, anchor with the terminal residues and add the three central residues. Peitsch M & Guex N (1997) Electrophoresis 18: 2714
Swiss-Model steps: Search for sequence similarities Evaluate suitable templates Generate structural alignments Average backbones Build loops Bridge incomplete backbones Rebuild sidechains Rebuild sidechains from rotamer library - complete sidechains first, then regenerate partial sidechains from probabilistic approach. Peitsch M & Guex N (1997) Electrophoresis 18: 2714
Swiss-Model steps: Search for sequence similarities Evaluate suitable templates Generate structural alignments Average backbones Build loops Bridge incomplete backbones Rebuild sidechains Energy minimize Gromos 96 - Energy minimization Peitsch M & Guex N (1997) Electrophoresis 18: 2714
Swiss-Model steps: Search for sequence similarities Evaluate suitable templates Generate structural alignments Average backbones Build loops Bridge incomplete backbones Rebuild sidechains Energy minimize Write Alignment and PDB file e-mail results Peitsch M & Guex N (1997) Electrophoresis 18: 2714
CASP5 (2002) - Homology better worse than template shocking! RMSD(target,template) – RMSD(target, model), Å Remote sequence similarity detection methods have improved. Coordinate manipulations do not improve accuracy. Tramontano A & Morea V (2003) Assessment of homology based predictions in CASP5 ProteinsS6:352-368
Swissmodel in comparison 3D-Crunch: 211,000 sequences -> 64,000 models Controls: >50 % ID: ~ 1 Å RMSD 40-49% ID: 63% < 3Å 25-29% ID: 49% < 4Å Manual alternatives: Modeller ... Automatic alternatives: SwissModel sdsc1 3djigsaw pcomb_pcons cphmodels easypred # 1 for RMSD and % correct aligned, #2 for coverage Guex et al. (1999) TIBS24:365-367 EVA:Eyrich et al. (2001) Bioinformatics17:1242-1243 (http://cubic.bioc.columbia.edu/eva)
Concept 4: SwissModel in practice.
SwissModel ... first approach mode http://www.expasy.org/swissmod
... run in Normal Mode (Except if defining a DeepView project )...
... successful submission. Results come by e-mail.
Homology Modeling in Practice How to assess model reliability ? - All indels are wrong - Structure analysis ("threading", "solvent accessibility", compatibility with ligands) can point out possible alignment errors - But: no point in "repairing" stereochemistry, only review alignment.
Homology Modeling in Practice Can you predict function from your model ? No (and yes) - the model may be incompatible with a specific function.
Uses of structure revisited - I: • Prototype 1: Analytical • Explain mechanistic aspects of protein. • (e.g. in terms of) • residues involved in catalysis • global properties (like electrostatics) • shape, relative orientation and distances of domains or subdomains • flexibility and dynamics - e.g. hypothesizing about the rate limiting step
Uses of structure revisited - II: • Prototype 2: Comparative • Bring conservation patterns into a spatial context in order to infer causality from (database) correlations. • (e.g. in terms of) • describing context specific conservation patterns and anlyizing these according to conserved properties • analyizing the predicted effect of sequence variation (e.g. for engineering changes, fusing domains or predicting SNP effects) • distinguish physiological vs. nonphysiological interactions
Questions ? Feedback ? boris.steipe@utoronto.ca http://biochemistry.utoronto.ca/steipe/