Download
1 / 11
110 likes | 410 Views
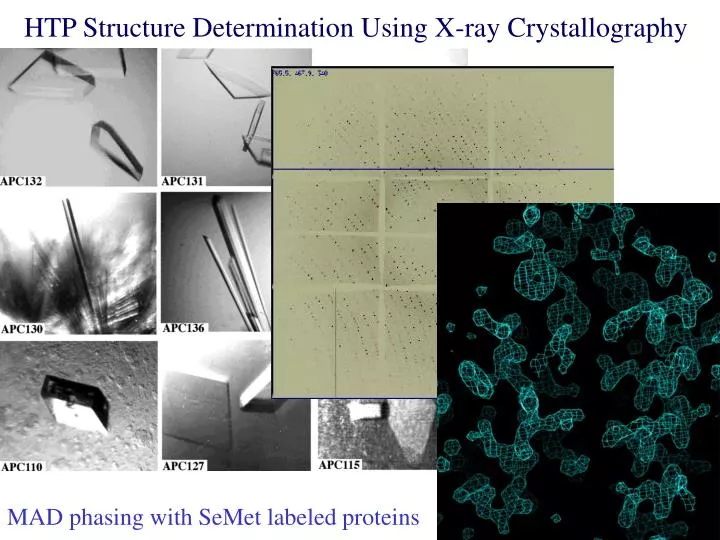
HTP Structure Determination Using X-ray Crystallography. MAD phasing with SeMet labeled proteins. What is Needed for HTP Structure Determination Using X-ray Crystallography?. X-ray quality mono-crystal Size >50x50x50 μm 3 Diffraction limit <3 Å

E N D
HTP Structure Determination Using X-ray Crystallography MAD phasing with SeMet labeled proteins
What is Needed for HTP Structure Determination Using X-ray Crystallography? • X-ray quality mono-crystal • Size >50x50x50 μm3 • Diffraction limit <3 Å • Ordered “heavy atom” (Se, Br, U, Hg, Xe, La, etc) for MAD analysis • on average 1 “heavy atom”/50-150 residues • Cryo-protection • “Crystallization grade” protein must be produced in quantities that permit: • Achieving protein concentration in the range 5-25 mg/ml • Testing 2x102 - 5x102 crystallization conditions • Growing X-ray quality mono-crystals • Producing derivative of similar quality and quantity that contains a “heavy atom” for structure determination • Establishing cryo-conditions
“Crystallization Grade” Quality #1 • Folded and soluble • Chemically and functionally homogeneous • As compact as possible • Flexible regions and affinity tags issues • In quantity and concentration suitable for crystallization experiments • Suitable for incorporation of a rational “heavy atom” for structure determination
“Crystallization Grade” Quality #2 • Free of critical contaminants that: • degrade, denature, destabilize or modify protein • interfere with crystallization • interfere with structure determination • Stable during crystallization trails • Must be functionally relevant
How to Get “Crystallization Grade” Protein? • Make your choice wisely – purification, concentration, storage and handling procedures play a critical role in obtaining “crystallization grade” protein samples • Quality of the protein sample depends on the method of preparation • Purification procedure should rapidly produce pure, homogeneous sample, however, a complete purity is not an absolute requirement for initial crystallization • Protein crystals are often obtained from mixtures • Crystallization was used to purify protein samples • Purification must be highly reproducible to supply milligram quantities of protein and its derivative containing “heavy atom”
Post-Purification Handling of Protein Samples • Protein concentration method • Short and long term storage conditions • Cross-contaminations • Protein denaturation and loses • During freezing • On solvent/air interface • Due to binding to surfaces during concentration and storage • Due to aggregation • Screening for metal-binding and other ligands
Characterization of Proteins for X-ray Crystallography • UV/VIS spectroscopy • Polyacrylamide gel electrophoresis (1D, 2D, N) • Dynamic and static light scattering • Circular dichroism spectroscopy • Mass spectroscopy • HSQC NMR spectroscopy • X-ray spectroscopy • Crystallization screens
“Crystallization Grade” Quality Membrane Proteins • Requirements for membrane proteins are, in general, similar to soluble proteins, but present a number of specific challenges: • Protein denaturation and long-term stability in detergents, detergent exchange procedures • Quality of detergents • Reconstitution of proteins into membrane environments • Removing affinity tags in the presence of detergents • Introduction of “heavy atom(s)” into membrane proteins
Challenges of Protein Sample Preparation Specific to HTP and Structural Genomics Projects • Large and diverse set of proteins: • From all three kingdoms of life • With low or no sequence similarity • With poorly characterized properties spanning a very wide range • Developing HTP universal procedures: “one size fits all” • Limited number of expression host strains and vectors • Widespread presence of codons infrequently used by the host • Host protein folding and assembly machinery is insufficient for folding of foreign proteins • A specific cofactor synthesis/import or post-translational modification function is missing in the host • Co-expression of a key interacting partner is missing • Errors in ORF annotation, domain assignment, sequencing etc • Introduction of heavy atom marker in vivo/in vitro is inefficient
In some cases gene/protein sequence errors do not seemto cause a problem Sequence Errors Ser Arg AGU CGU His Ile CAU AUU
Challenges of Protein Sample Preparation Specific to HTP and Structural Genomics Projects • Large scale requires parallel processing of multiple protein samples throughout all steps • Use LIMS to develop “database” driven procedures: • Maintaining large databases in “real time” • Database mining to improve/optimize methods and procedures • Assuring public access • Low cost